
Nesarkomerické formy hypertrofické kardiomyopatie v dospělosti
Non-sarcomeric hypertrophic cardiomyopathies in adults
Hypertrophic cardiomyopathy is the most common cardiovascular disease of genetic origin. In the majority of cases it is an autosomal dominant hereditary disease marked by mutation of one of the genes for sarcomeric proteins and so-called sarcomeric hypertrophic cardiomyopathy. In approximately one third of patients, however, it is not possible to prove the cause of the mutation in the gene responsible for coding the sarcomeric protein. Apart from the hitherto undescribed sarcomeric mutations, the hardening of the walls of the myocardium could originate in a different genetic abnormality, the substance of which is, in the majority of cases, an enzymatic dysfunction at some level of cellular metabolism. These forms of hypertrophic cardiomyopathy are generally known as non-sarcomeric phenocopies. The authors of this report give an overview of the most frequent types of non-sarcomeric hypertrophic cardiomyopathy found in adults. A detailed study is made of the issue of Fabry disease (FCH), Danon disease (DCH), PRKAG2 syndrome, mitochondrial hypertrophic cardiomyopathy and cardiomyopathy during Friedreich’s ataxia.
Keywords:
hypertrophic cardiomyopathy – non-sarcomeric phenocopy – Fabry disease – Danon disease – PRKAG2 syndrome – mitochondrial disorders – Friedreich’s ataxia
Autoři:
T. Paleček 1; P. Kuchynka 1; E. Němeček 1; M. Mašek 2; M. Elleder 3; T. Honzík 4; A. Linhart 1
Působiště autorů:
II. Interní klinika – klinika kardiologie a angiologie, 1. LF UK v Praze a VFN v Praze
1; Radiodiagnostická klinika, 1. LF K v Praze a VFN v Praze
2; Ústav dědičných metabolických poruch, 1. LF UK v Praze a VFN v Praze
3; Klinika dětského a dorostového lékařství, 1. LF UK v Praze a VFN v Praze
4
Vyšlo v časopise:
Kardiol Rev Int Med 2011, 13(4): 210-220
Souhrn
Hypertrofická kardiomyopatie je nejčastějším geneticky podmíněným kardiovaskulárním onemocněním. Ve většině případů se jedná o autozomálně dominantně dědičnou chorobu podmíněnou mutací v některém z genů pro sarkomerické proteiny, tzv. sarkomerickou hypertrofickou kardiomyopatii. U přibližně jedné třetiny nemocných však není možné příčinnou mutaci v genu kódujícím sarkomerický protein prokázat. Kromě dosud nepopsaných sarkomerických mutací může být podkladem zesílení stěn myokardu jiná geneticky podmíněná odchylka, jejíž podstatou je ve většině případů enzymatická dysfunkce na některé úrovni buněčného metabolizmu. Tyto formy hypertrofické kardiomyopatie jsou obecně nazývány nesarkomerické fenokopie. Autoři sdělení podávají přehled nejčastějších typů nesarkomerických hypertrofických kardiomyopatií, s nimiž se lze setkat v dospělosti. Podrobně je rozvedena problematika Fabryho choroby (FCH), Danonovy choroby (DCH), PRKAG2 syndromu, mitochondriální hypertrofické kardiomyopatie a kardiomyopatie při Friedreichově ataxii.
Klíčová slova:
hypertrofická kardiomyopatie – nesarkomerická fenokopie – Fabryho choroba – Danonova choroba – PRKAG2 syndrom – mitochondriopatie – Friedreichova ataxie
Úvod
Hypertrofická kardiomyopatie je definována přítomností zvýšené tloušťky stěny či celkové hmotnosti levé komory srdeční (LK) při absenci takového stupně zatížení podmíněného arteriální hypertenzí či chlopenní vadou, které by mohlo danou abnormalitu myokardu vyvolat [1]. Její prevalence dosahuje dle současných znalostí 1 : 500, a je tak nejčastější geneticky podmíněnou kardiovaskulární chorobou [2]. Jedná se o onemocnění se značnou fenotypickou a genotypickou heterogenitou. Ve většině případů jsou genetickým podkladem mutace v genech kódujících strukturu sarkomerických proteinů a přenos choroby je typicky autozomálně dominantní [3]. Tehdy hovoříme o tzv. sarkomerické hypertrofické kardiomyopatii. Extenzivní genetický screening ale nedokáže příčinnou mutaci genu sarkomerického proteinu identifikovat u 30–40 % jedinců [4]. Zde se může jednat buď o sarkomerickou kardiomyopatii na podkladě dosud nepoznané a nepopsané mutace, nebo o tzv. nesarkomerickou formu hypertrofické kardiomyopatie, která je také podmíněna geneticky, ale mutace postihuje gen pro jiné než sarkomerické proteiny. Cílem tohoto sdělení je podat přehled nejčastějších typů nesarkomerických kardiomyopatií, s nimiž se lze setkat v dospělosti. Podrobně je rozvedena problematika Fabryho choroby (FCH), Danonovy choroby (DCH), PRKAG2 syndromu, mitochondriální hypertrofické kardiomyopatie a kardiomyopatie při Friedreichově ataxii. V současnosti lze mezi nesarkomerické formy hypertrofické kardiomyopatie zařadit i srdeční amyloidózu ve stadiu ještě nerestriktivního postižení diastolické funkce LK [1]; problematice amyloidotické kardiomyopatie je v tomto čísle věnován samostatný článek.
Fabryho choroba
Fabryho choroba patří do skupiny vrozených střádavých lyzozomálních onemocnění, glykosfingolipidóz. Základním genetickým defektem je na chromozom X vázaná porucha aktivity lyzozomálního enzymu α-galaktozidázy A s následnou intracelulární akumulací neutrálních glykosfingolipidů, především globotriaosylceramidu, v řadě tkání a orgánových systémů [5]. U hemizygotních mužů s klasickým fenotypem FCH je aktivita α-galaktozidázy A nulová nebo velmi nízká. U žen, které byly dříve ne zcela správně označovány jen jako přenašečky, je aktivita enzymu v průměru poloviční, ojediněle může dosahovat i zcela normálních hodnot či naopak se blížit nule. Příčinou postižení heterozygotních žen je náhodná inaktivace jednoho ze dvou chromozomů X, která u části buněk ponechává aktivní deficitní gen pro α-galaktozidázu A (tzv. lyonizace) [6]. Výskyt manifestací FCH je u žen ve srovnání s muži méně intenzivní a zpravidla je asi o 10 let opožděn [7].
Jedná se o onemocnění panetnické, s odhadovanou incidencí 1 : 40 000 až 1 : 117 000 narozených mužů [8]. Dosud bylo popsáno více než 300 mutací genu α-galaktozidázy A. Každá mutace je prakticky specifická (privátní) pro jednu, maximálně několik málo postižených rodin [9]. Více než polovinu známých mutací představují jednoduché „missense“ mutace, které vedou k záměně jedné aminokyseliny za druhou.
Fabryho choroba je metabolickou poruchou s pomalou progresí, bez infaustních fatálních forem. První klinické manifestace se obvykle objevují již v dětství a v dospívání. Jedná se o akroparestezie, což jsou neuropatické bolesti postihující dlaně a plosky, dále o febrilní krize, hypohidrózu a s tím související intoleranci tepla, gastrointestinální potíže a postupný výsev specifických kožních lézí, tzv. angiokeratomů. V časné dospělosti se objevuje proteinurie a neurologické manifestace zahrnující poruchy sluchu a vestibulárního aparátu [5]. Kardiální postižení je klinicky detekovatelné až v třetí dekádě života a jeho primárním podkladem je depozice globotriaosylceramidu v kardiomyocytech, buňkách převodního systému, chlopenních fibroblastech a endoteliálních a svalových buňkách cév [10]. Velmi zřídka je u některých mužů přítomna určitá reziduální aktivita α-galaktozidázy A, což je podkladem tzv. kardiální varianty onemocnění, kdy u postižených jedinců chybí zjevné extrakardiální manifestace choroby a klinickému obrazu dominuje myokardiální postižení [11]. Rigorózní diagnostika ovšem i u těchto nemocných nalézá subklinické postižení jiných orgánů, především mikroalbuminurii, a proto je existence izolované kardiální formy FCH zpochybňována.
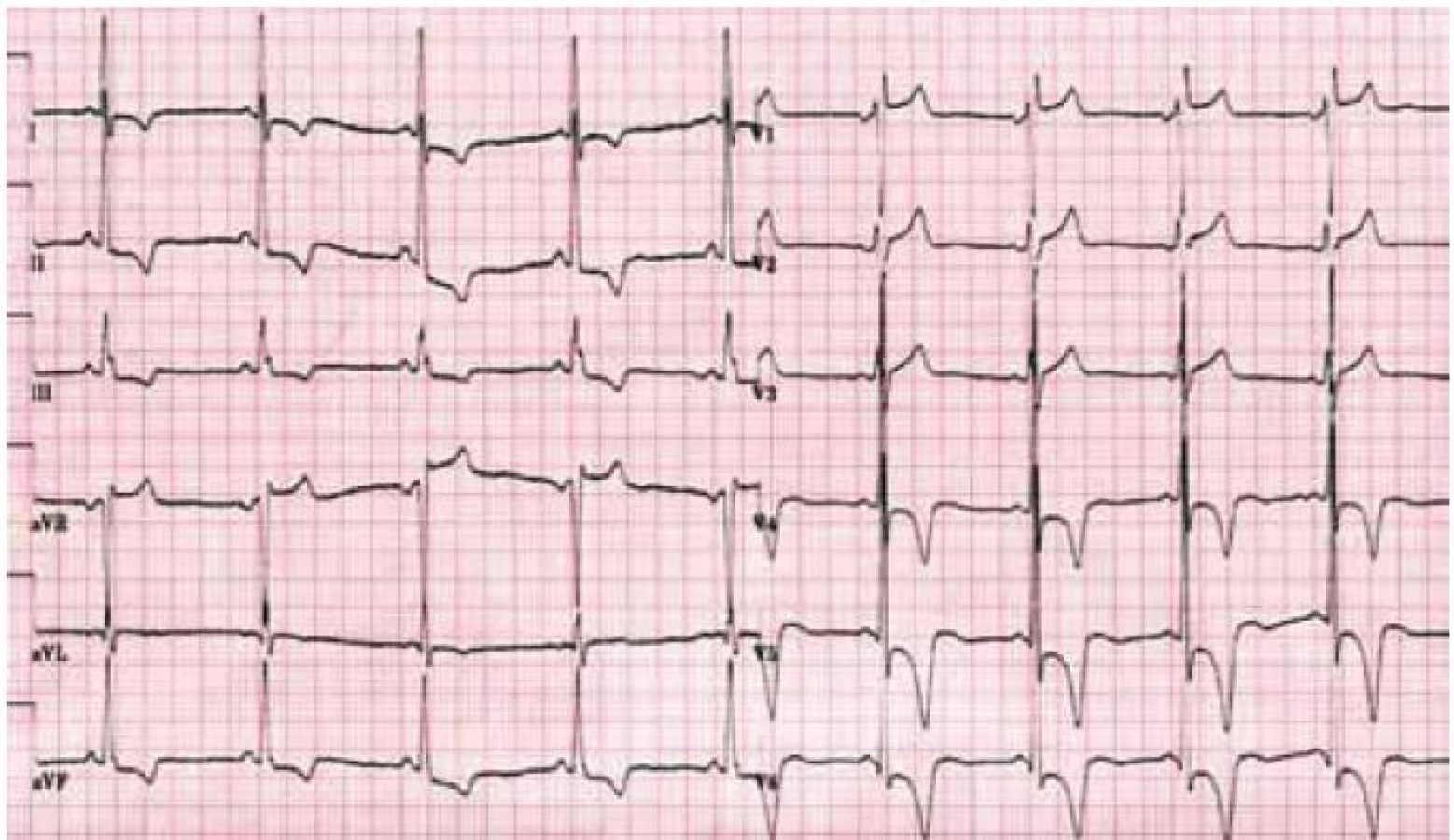
Hlavní kardiální manifestací u FCH je vývoj hypertrofie LK. V časné fázi dochází ke koncentrické remodelaci komory, která postupně progreduje do typického obrazu koncentrické hypertrofie (obr. 1) [12]. Asymetrická septální hypertrofie, která dominuje fenotypickému obrazu sarkomerické HKMP, je přítomna asi u 5 % nemocných. Vzácně lze nalézt dynamickou obstrukci výtokového traktu LK. Celková systolická funkce LK je dle konvenčních parametrů, jako je ejekční frakce, dlouhou dobu zachována, nicméně techniky hodnotící regionální deformaci myokardu prokazují u kardiomyopatie spojené s FCH snížení nejprve longitudinální a posléze i radiální systolické funkce [13]. Diastolická funkce hypertrofické LK je pochopitelně postižena, ale většinou jen mírně až středně těžce [12]. Těžký restriktivní typ diastolické dysfunkce LK je přítomen vzácně a objevuje se až v konečných fázích těžkého myokardiálního postižení, které je též spojeno s postupným poklesem celkové systolické funkce LK [14]. Zároveň s vývojem hypertrofie LK dochází také k zesílení stěn pravé komory, jejíž systolická funkce je normální a vyjádřen je opět většinou jen mírný až střední stupeň diastolické dysfunkce [15]. Postižení chlopenního aparátu je u FCH charakterizováno zesílením cípů aortální a mitrální chlopně, které však u většiny nemocných nevede k hemodynamicky významným valvulopatiím [12]. Na EKG jsou typicky přítomny voltážové známky hypertrofie LK s doprovodnými změnami v repolarizaci. U mladých jedinců je velmi často přítomno zkrácení PQ intervalu, které je podmíněno urychlením atrioventrikulárního převodu, nikoli akcesorní spojkou (obr. 2) [16]. S progredujícím postižením převodního systému dochází u starších nemocných k dysfunkci sinusového uzlu, prodlužování PQ intervalu až do obrazu atrioventrikulárních blokád různého stupně a vývoji raménkových blokád. Supraventrikulární tachyarytmie, především fibrilace síní, se vyskytují u téměř jedné pětiny jedinců s FCH [17]. Nesetrvalé komorové tachykardie lze pomocí Holterova monitorování dokumentovat u cca 8 % nemocných, náhlá srdeční smrt však není pro FCH typickou [18].
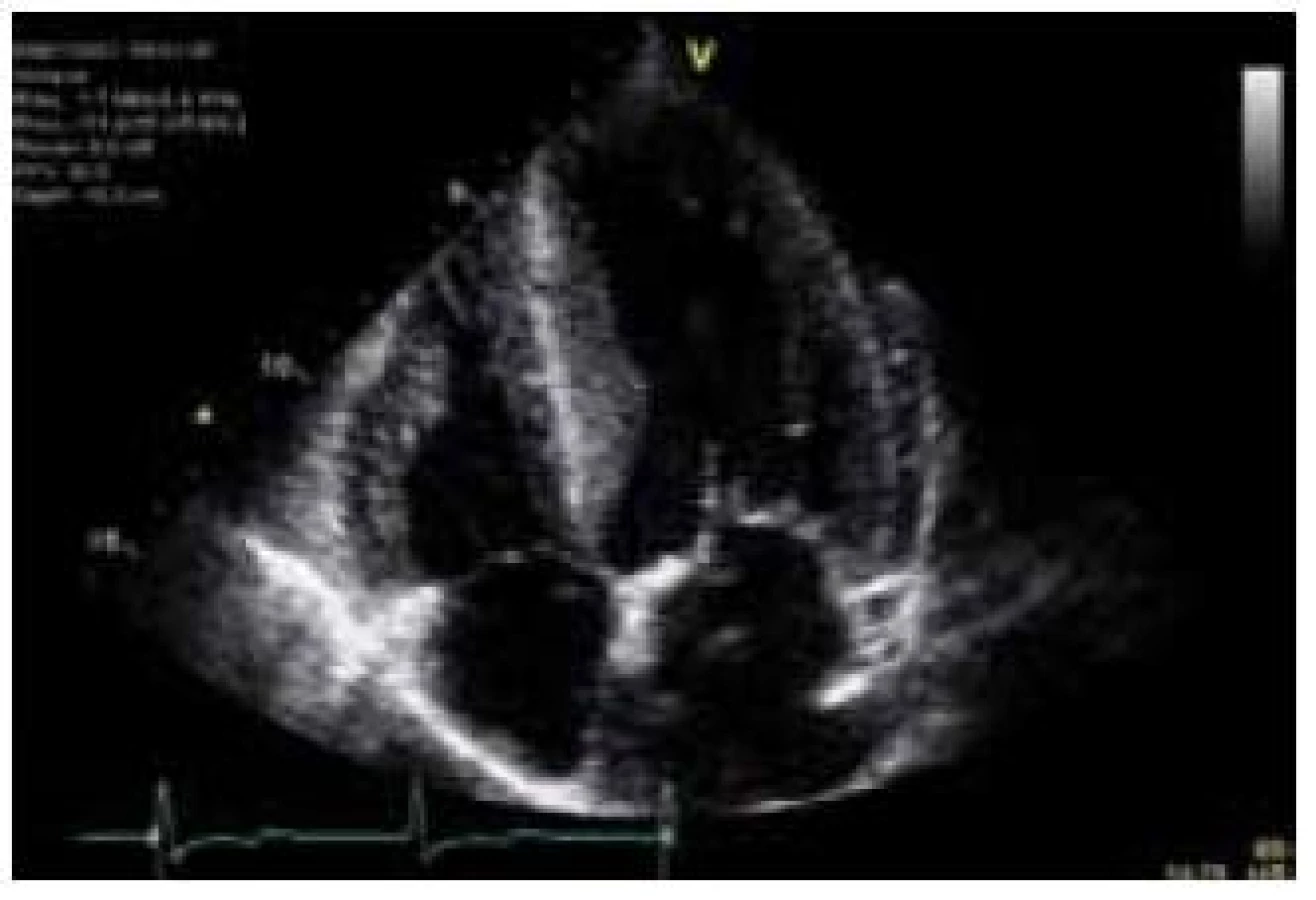
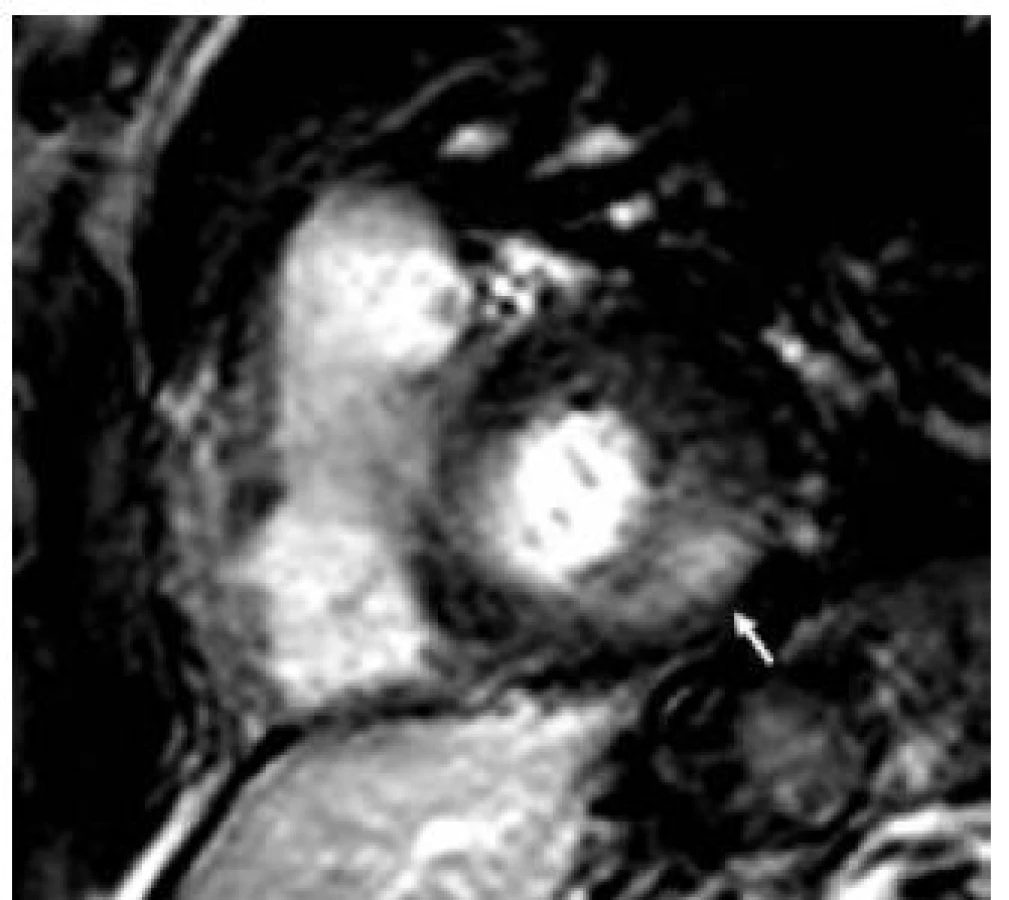
Patofyziologie vývoje hypertrofické kardiomyopatie při FCH není doposud zcela jasně objasněna, ale lze ji pokládat za obecnou reakci srdečního svalu nejen na lyzozomální střádání, ale i např. na depozici glykogenu. V případě FCH totiž představuje depozice globotriaosylceramidu pouze 1–2 % celkové masy myokardu, dominantní složkou je vlastní hypertrofie svalových vláken [19]. K té pravděpodobně dochází v důsledku působení zatím ne zcela přesně identifikovaných růstových faktorů přítomných v plazmě jedinců s FCH. Jedním z nich se jeví být globotriaosylsfingozin, lysoderivát globotriaosylceramidu [20]. Svou roli při vývoji hypertrofie myokardu, ať již primární či sekundární, jistě sehrává i porucha energetického metabolizmu [21]. Malou, nicméně funkčně důležitou část celkové myokardiální masy představuje u kardiomyopatie při FCH intersticiální fibróza. Její přítomnost lze klinicky nejlépe detekovat pomocí pozdního sycení kontrastní látkou při vyšetření magnetickou rezonancí [22]. Zcela typickou lokalizací pozdního sycení kontrastní látkou u FCH je midmyokardiální oblast bazálního posterolaterálního segmentu LK (obr. 3) [23]. V souvislosti s tím dochází v tomto regionu u pokročilých forem myokardiálního postižení k zeslabování a zhoršování kinetiky stěny komory [24].
Diagnóza FCH je u hemizygotních mužů velmi jednoduchá a spočívá ve stanovení aktivity α-galaktozidázy A v plazmě, periferních leukocytech a fibroblastech. Velmi přínosná je i analýza močových lipidů, vzhledem k tomu, že ledvinné glomeruly i tubuly jsou postiženy střádáním. Toto vyšetření je symbolicky nazýváno „chemickou biopsií ledvin“. Definitivní průkaz diagnózy FCH ovšem dává až průkaz přítomnosti specifické rodinné mutace na příslušném genu. Genetická diagnostika je, kromě bioptického vyšetření postižených orgánů, jedinou možností u heterozygotních žen, které mohou mít normální až velmi nízkou hladinu α-galaktozidázy A [25]. Rovněž výše uvedená „chemická biopsie ledvin“ má u žen menší výpovědní hodnotu. Významným pokrokem v diagnostice FCH, umožňujícím plošný screening v rizikových skupinách nemocných, bylo zavedení vyšetření aktivity α-galaktozidázy A pomocí testu suché krevní kapky na filtračním papíře [26]. Diagnostika FCH je v České republice prováděna v Ústavu dědičných metabolických poruch 1. LF UK v Praze.
Prevalencí FCH u jedinců s hemodynamicky nevysvětlitelnou hypertrofií LK se zabývala již řada prací, které prokazovaly její přítomnost u 0–12 % nemocných s fenotypickým obrazem hypertrofické kardiomyopatie [27–30]. Nejvalidnější studií je v tomto ohledu patrně práce Monseratta et al, kteří vyšetřili největší soubor 508 nemocných s hypertrofickou kardiomyopatií, sledovaných v terciárních centrech ve Španělsku, a geneticky verifikovanou diagnózu FCH zjistili u tří mužů a dvou žen, tedy u 1 % zkoumané populace [31]. K podobnému závěru spěje i česká prospektivní screeningová studie přítomnosti FCH u mužů s nevysvětlitelnou hypertrofií LK, sledovaných primárními kardiology, do níž je plánováno zařadit celkem 100 nemocných [32]. V rámci této studie bylo od roku 2005 vyšetřeno pomocí testu suché krevní kapky či klasickým stanovením aktivity α-galaktozidázy A v plazmě již 85 jedinců a FCH byla potvrzena u dvou z nich [33].
Základním kamenem léčby FCH včetně kardiálních manifestací je v současnosti enzymatická substituční terapie. K dispozici jsou dvě formy rekombinantního enzymu: agalsidáza α, která je získávána z kultur lidských kožních fibroblastů s aktivovanou produkcí enzymu, a agalsidáza β, kdy je enzym derivován ze standardní linie ovariálních buněk čínských křečků. U obou preparátů prokázala většina dosud publikovaných observačních studií, provedených na malých kohortách nemocných, statisticky významnou regresi hmotnosti LK hodnocenou jak echokardiograficky, tak pomocí magnetické rezonance [34]. Nedávná studie českých autorů opřená o několikaleté bioptické sledování však ukázala velmi omezenou efektivitu enzymatické substituční terapie, zejména pokud jde o pozitivní vliv na myokard [35]. Též prospektivní studie Koskenvuoa et al, zahrnující devět nemocných s FCH, neprokázala pozitivní vliv enzymatické substituční terapie podávané po dobu 24 měsíců na morfologii a funkci LK [36]. Omezený efekt enzymatické substituční terapie je velmi pravděpodobně podmíněn pozdním nástupem léčby, modifikací střádacích lyzozomů a skutečností, že kriticky postiženými buňkami jsou kardiomyocyty a glomerulární podocyty, které nepodléhají obměně. Tuto hypotézu potvrzuje i studie Weidemanna et al, v níž byl efekt specifické léčby závislý na pokročilosti myokardiálního postižení, konkrétně na tíži hypertrofie LK a přítomnosti ložisek fibrózy detekovaných magnetickou rezonancí [37]. Obecně je tedy důležité zahájit specifickou léčbu ještě v časných stadiích onemocnění, před vývojem ireverzibilních změn. Hlavní limitací takovéto enzymatické „preventivní“ terapie je však její ekonomická náročnost. Diagnostikou subklinického postižení myokardu u FCH se zabývaly dvě práce německých a italských autorů, ve kterých byla demonstrována možnost detekce preklinických abnormalit diastolické funkce a regionální systolické funkce pomocí tkáňové dopplerovské echokardiografie a deformační analýzy ještě před vývojem hypertrofie LK [38,39].
Kromě enzymatické substituční terapie je nemocným s kardiálními manifestacemi FCH podávána i konvenční léčba, jejíž benefit byl prokázán u jiných srdečních onemocnění. Při známkách srdečního selhávání jsou podávány ACE inhibitory, diuretika a spironolakton, betablokátory pak s velkou opatrností vzhledem k možnosti potenciace poruchy převodního systému. Podávání ACE inhibitorů či sartanů je vhodné i vzhledem k renálnímu postižení a proteinurii. Pacienti s těžkým stupněm srdečního selhávání mohou být kandidáti transplantace srdce, kdy vlastní produkce enzymu transplantovaným srdcem by měla zabránit rekurenci srdečního postižení [17]. U řady nemocných je pro progredující poruchu převodního systému nutná implantace trvalého kardiostimulátoru. Antikoagulace je plně indikována při záchytu fibrilace či flutteru síní. Implantace kardioverteru-defibrilátoru se řídí obecnými indikačními pravidly pro jedince s kardiomyopatiemi. Vzácné případy dynamické obstrukce výtokového traktu LK lze efektivně řešit perkutánní alkoholovou septální ablací [40].
Danonova choroba
Danonova choroba je vzácnou lyzozomální poruchou, která se vymyká klasickým lyzozomálním střádacím onemocněním, jejichž příčinou je enzymový deficit. U DCH jde o deficit nekatalytického lyzozomálního membránového proteinu 2 (LAMP-2), který je kódován na chromozomu X [41]. Tento protein je vysoce glykosylován. Jeho funkce není zatím kompletně objasněna. Existují důkazy, že působí jako transmembránový transportér pro denaturované proteiny, které mají být do lyzozomu importovány [42]. Vedle toho se předpokládá i významná role v transformaci autofagozomu, který je neschopen degradace, na autofagolyzozom schopný degradace. To vysvětluje, proč byla DCH původně nazývána „lyzozomální glykogenózou s normální aktivitou kyselé α-glukozidázy“, neboť v bioptických vzorcích postižených tkání byly v cytoplazmě nalézány autofagické vakuoly s množstvím granul glykogenu, aniž by byl prokazatelný deficit kyselé α-glukozidázy, což odlišuje DCH od Pompeho nemoci, glykogenózy typu II [41].
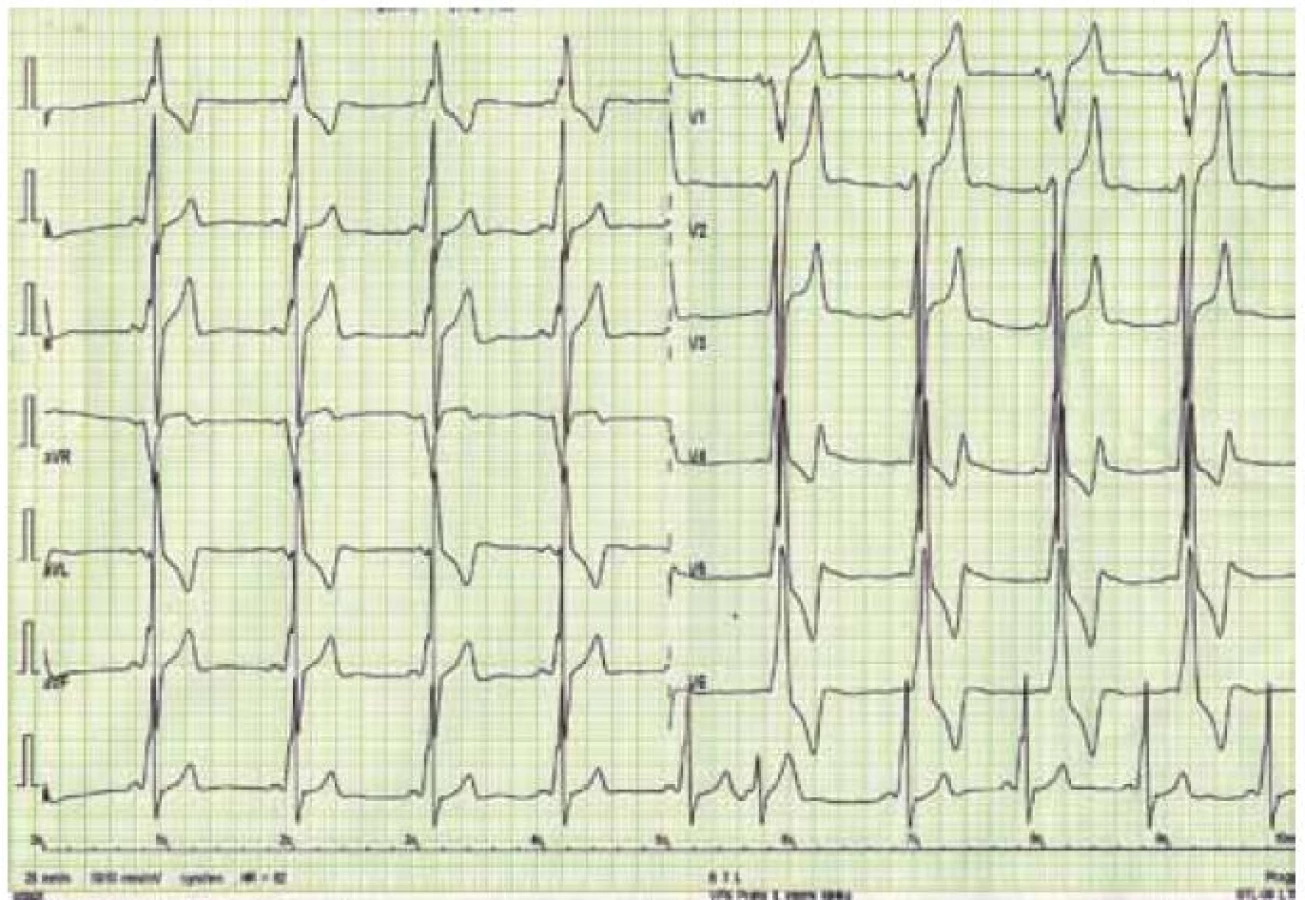
Kromě několika kazuistických sdělení byly dosud publikovány jen dvě velké práce zabývající se manifestacemi a průběhem DCH na větších souborech nemocných, čítajících 20 mužů a 18 žen z 13 rodin v první z nich, resp. 43 mužů a 39 žen z 36 rodin v druhé [43,44]. Klasická fenotypická triáda DCH je charakterizována kardiomyopatií, skeletální myopatií a mentální retardací [45]. Klinický průběh je u mužů charakterizován časným počátkem a velmi špatnou prognózou. Známky kardiálního postižení se u nich nejčastěji objevují mezi 10. až 15. rokem života, ale vždy do 20 let věku. Typicky dochází k rozvoji symetrického zesílení stěn LK, které může dosahovat až excesivních hodnot [46]. Ejekční frakce LK je iniciálně zachována, brzy však dochází k jejímu zhoršování až do obrazu těžké celkové systolické dysfunkce, doprovázeného postupnou dilatací LK [47]. Dynamická obstrukce výtokového traktu LK byla popsána jen v zcela ojedinělých případech [30]. Tloušťka stěny pravé komory bývá také výrazně zesílena. Na EKG je u většiny nemocných přítomen typický obraz preexcitace s krátkým PQ intervalem, často vyjádřenou delta vlnou a voltážové známky hypertrofie LK (obr. 4) [46]. Urychlení atrioventrikulárního převodu je u DCH přikládáno existenci pravých akcesorních spojek podmíněných narušením fibrózního anulu kardiomyocyty naplněnými glykogenem [48]. Poruchy převodního systému ve smyslu AV blokád byly ovšem též u některých mužů s DCH popsány, podobně jako různé druhy supraventrikulárních arytmií [45]. Pozdní sycení kontrastní látkou při vyšetření magnetickou rezonancí vykazuje subendokardiální až midmyokardiální, místy téměř transmurální charakter a typicky postihuje volné stěny a hrot LK (obr. 5) [49]. Skeletální myopatie bývá u většiny nemocných mírného stupně, někdy je klinicky inaparentní, ale i v těchto případech lze nalézt zvýšené sérové hladiny kreatinkinázy [30]. Určitý stupeň mentální retardace je také vyjádřen u většiny postižených jedinců, jeho absence, stejně jako nepřítomnost zjevné myopatie, však přítomnost DCH nevylučuje [30,43]. Jelikož deficience LAMP-2 postihuje všechny tělesné buňky, je u některých nemocných přítomno i postižení očí (sítnice), jater a dalších orgánů [50]. Jak bylo uvedeno výše, DCH má u mužů dramaticky progresivní charakter. Dle literárních údajů zemřeli všichni dosud diagnostikovaní muži, kromě dvou případů, v třetím deceniu, a to na neovlivnitelné srdeční selhání či náhlou srdeční smrt způsobenou maligní komorovou arytmií [44,47].

U žen dochází k manifestaci onemocnění až v dospělosti a průběh choroby je u nich obecně méně agresivní. Skeletální myopatie a poruchy intelektu jsou méně časté a mírnějšího stupně, často málo klinicky vyjádřené a nezřídka zcela absentují [43]. Prevalence výskytu symptomatického srdečního postižení je ovšem dle recentní studie autorů Boucek et al poměrně vysoká [44]. Na rozdíl od dřívější představy, že dominantní formu kardiálního fenotypu představuje u žen dilatační kardiomyopatie, ukazuje tato práce na téměř rovnoměrné riziko vývoje hypertrofické i dilatační kardiomyopatie.
Otázkou, jaká je prevalence DCH v populaci dospělých jedinců s obrazem hypertrofické kardiomyopatie, se dosud zabývaly dvě práce Charrona et al a Arada et al, které zjistily přítomnost mutace pro LAMP-2 u 1, resp. 4 % zkoumaného souboru [30,45]. K obdobnému závěru dospěla i pediatrická screeningová studie amerických autorů, která prokázala DCH u 4 % dětských jedinců s hypertrofickou kardiomyopatií [51].
Diagnostika DCH je u mužů založena na charakteristickém nálezu v bioptickém vzorku z periferního svalu či myokardu, kde kromě výše uvedených známek vakuolární myopatie je imunohistochemicky prokázána absence LAMP-2 [45]. Italskými autory byla ukázána možnost neinvazivního průkazu nepřítomnosti LAMP-2 v leukocytech periferní krve (v systému lyzozomům blízkých organel), což významně rozšiřuje diagnostické možnosti DCH především ve smyslu širšího screeningu její přítomnosti u mužů s hypertrofickou kardiomyopatií [50]. Toto vyšetření bylo v nedávné době zavedeno ve standardních nátěrech periferní krve i v Ústavu dědičných metabolických poruch 1. LF UK v Praze [52]. Definitivně je ovšem diagnóza stanovena geneticky, průkazem specifické mutace v genu pro LAMP-2, což je též jediná možnost diagnostiky DCH u žen.
V současné době není k dispozici specifická léčba DCH. Vzhledem k výrazné malignitě kardiálního postižení, charakterizované náhle vzniklou a rychle progredující deteriorací systolické funkce LK s vysokou tendencí k maligním komorovým arytmiím a ústící do terminálního srdečního selhání, je na místě extrémně pečlivé a frekventní sledování postižených jedinců. Otázkou je efektivita primárně preventivní implantace kardioverteru-defibrilátoru, neboť v recentní Maronově sérii nebyla letální arytmie defibrilátorem terminována u pěti ze sedmi nemocných s kardiomyopatií při DCH [47]. Důležitá je včasná indikace k transplantaci srdce, nicméně vzhledem k letalitě choroby bylo takto léčeno jen několik jedinců [47,53].
PRKAG2 syndrom
Mutace v genu PRKAG2 na sedmém chromozomu, kódujícímu γ-podjednotku adenozin-monofosfátem aktivované proteinkinázy (AMPK), jsou podkladem jiné nesarkomerické hypertrofické kardiomyopatie asociované s elektrofyziologickými abnormalitami [54]. AMPK působí jako buněčný „energetický senzor“, který je aktivován při depleci ATP a indukuje řadu následných pochodů vedoucích k efektivnímu využití energie [55]. Jedním z těchto kroků je potenciace transportu glukózy do myocytu a syntéza glykogenu, jehož zmnožení může simulovat mírnou formu glykogenózy. Jedná se tedy o proces zvýšené syntézy glykogenu, nikoli jeho nedostatečné degradace jako u glykogenóz. Podle některých sdělení může paralelně docházet i k depozici amylopektinu, nejspíše tím, že lineární syntéza katalyzovaná glykogensyntázou není sledována dostatečně intenzivní aktivitou větvícího enzymu. Poruchu charakterizuje minimální stupeň intersticiální fibrózy, která je prokazatelná při bioptickém vyšetření [54]. Přenos choroby je autozomálně dominantní, s plnou penetrancí [56]. V České republice nebyla doposud diagnostikována.
Prevalence PRKAG2 kardiomyopatie v obecné populaci jedinců s HKMP je velmi nízká, nepřesahující zřejmě 1 % [54,57]. Proto byly zatím, podobně jako u DCH, publikovány jen dvě práce, studující větší soubory nemocných s geneticky prokázanou mutací PRKAG2, z nichž lze usuzovat na přirozený vývoj onemocnění (navíc jen jedna z nich, Murphyho et al, byla tímto směrem primárně zaměřena) [54,57]. K manifestaci klinických symptomů dochází nejčastěji ve věku kolem 25 let. Hypertrofie LK může mít různý charakter, koncentrický či asymetrický, a to i mezi členy jedné postižené rodiny. Její stupeň je středně až velmi těžký. Dynamická obstrukce výtokového traktu je zcela unikátní. Relativně častá je naopak těžká porucha diastolické funkce LK. S věkem dochází obvykle k progresi hypertrofie, která je u některých nemocných doprovázena dilatací a postupným snižováním globální systolické funkce LK, jež může dosáhnout výrazného stupně a vést k indikaci transplantace srdce. Přežívání jedinců s PRKAG2 kardiomyopatií je ale zřetelně delší než u nemocných s DCH. Zajímavou skutečností ovšem je, že byl popsán i případ rychle progredující infantilní hypertrofické kardiomyopatie, podmíněné stejnou genetickou abnormalitou, což ostře kontrastuje s průběhem onemocnění u dospělých jedinců [58].
Hlavní klinický problém přestavuje pro nemocné s mutací PRKAG2 progredující porucha převodního systému. Iniciálně vykazuje většina postižených jedinců na EKG obraz preexcitace se zkrácením PQ intervalu a přítomností delta vln. Podobně jako u DCH je podkladem urychleného atrioventrikulárního vedení přítomnost pravých akcesorních spojek pronikajících fibrózním anulem [48, 59]. Běžné jsou supraventrikulární tachykardie včetně fibrilace síní, které jsou spojeny s vyšším rizikem kardioembolické cévní mozkové příhody. Poměrně rychle však dochází se stárnutím individua ke zpomalení převodu vzruchu a vývoji bradyarytmií zahrnujících syndrom chorého sinu a různé formy atrioventrikulárních blokád, které u více než jedné třetiny nemocných vyžadují implantaci trvalého kardiostimulátoru. Komorové tachykardie a náhlá srdeční smrt jsou naopak vzácné, asociované s přítomností výrazné hypertrofie levé komory [57].
Ačkoli původní názory předpokládaly, že mutace PRKAG2 vede k izolované formě kardiální glykogenózy [60], v práci Murphyho et al byly klinické známky velmi lehké skeletální myopatie projevující se pozátěžovou myalgií popsány u 15 % nemocných [57]. U většiny takto postižených jedinců byl elektromyogram normální, histopatologické vyšetření však postižení periferních svalů potvrdilo.
Diagnóza PRKAG2 kardiomyopatie je založena na výsledku bioptického vyšetření vzorku myokardu a genetickém průkazu mutace genu PRKAG2. V České republice je diagnostika možná v Ústavu dědičných metabolických poruch 1. LF UK v Praze.
Specifická léčba tohoto syndromu není k dispozici a její vývoj je zcela v počátcích, na úrovni akademického experimentálního výzkumu [61]. Symptomy srdečního selhání jsou léčeny konvenční medikací diuretiky a ACE inhibitory. Betablokátory je nutné podávat s vysokou opatrností vzhledem k progredující dysfunkci převodního systému. Nutné je pravidelné Holterovské monitorování EKG a zahájení antikoagulační léčby při záchytu byť i paroxyzmální fibrilace síní. Implantace kardioverteru-defibrilátoru se řídí obecnými indikačními pravidly pro nemocné s hypertrofickou kardiomyopatií [57]. Při rychlé deterioraci systolické funkce LK je nutná včasná indikace k srdeční transplantaci.
Mitochondriální kardiomyopatie
Mitochondrie jsou dynamické organely, jejichž primární funkcí je podpora aerobní respirace a tvorba vysokoenergetických substrátů, jako je ATP [62]. Odehrává se v nich řada kruciálních metabolických procesů včetně Krebsova cyklu a β-oxidace mastných kyselin. Mají též důležitou úlohu v regulaci apoptotické buněčné smrti [63]. Hustota mitochondrií se v jednotlivých tkáních liší a je podmíněna jejich energetickými potřebami. Vysokou denzitu mitochondrií proto nalézáme v neuronech, myocytech a kardiomyocytech, což vysvětluje citlivost těchto buněk k poruchám tvorby energie podmíněných mitochondriopatiemi. Za mitochondriální poruchy jsou označovány genetické defekty pyruvátdehydrogenázového cyklu, Krebsova cyklu a respiračního řetězce. Proteiny, které se v mitochondriích účastní metabolických procesů, jsou kódovány jednak nukleární DNA, jednak vlastní mitochondriální DNA (mtDNA), která se v každé mitochondrii nachází v cca 10–20 kopiích a obsahuje genetickou informaci pro tvorbu proteinů zásadně se účastnících oxidativní fosforylace [62]. Dysfunkce mitochondrií může být proto podmíněna mutacemi jak v nukleární, tak i v mtDNA. Mutace mohou být bodové nebo se jedná o delece. Mitochondriální DNA vykazuje maternální typ dědičnosti, neboť mitochondrie, spolu s ostatními organelami, pocházejí výlučně z vajíčka. Během dělení zygoty dochází vždy znovu a znovu k náhodné distribuci normální a mutované mtDNA do dceřiných buněk. Výsledkem je neuniformní distribuce mtDNA jak v mitochondriích samotných, tak v buňkách a na orgánové úrovni. Na konci embryonální diferenciace individua pak nacházíme různý poměr normální a mutované mtDNA v jednotlivých tkáních a orgánech. Biochemicky lze tuto neuniformní distribuci stanovit poměrem mezi normální a mutovanou mtDNA, což se označuje jako heteroplazmie [62]. Fenotypové vyjádření daného defektu oxidativní fosforylace a respiračního řetězce je proto podmíněno vlastní biochemickou podstatou defektu, stupněm heteroplazmie v jednotlivých tkáních a orgánech, jejich energetickou náročností, věkem individua a jeho pohlavím [64]. Nukleárně podmíněné mitochondriální poruchy heteroplazmii postrádají. Orgánové postižení je tak uniformní a rozdíly jsou pouze meziorgánové. Toto vše vede k výrazné klinické heterogenitě mitochondriopatií ve smyslu manifestace onemocnění (od novorozeneckého věku až po dospělost), typu a stupně orgánového postižení.
Mitochondriální kardiomyopatie je možné definovat jako primární poruchy oxidativní fosforylace charakterizované abnormálním počtem, strukturou a/nebo funkcí mitochondrií srdečního svalu [65]. Většinou jsou kardiomyopatie jen jednou ze součástí multisystémového postižení. Systémy a orgány, které bývají kromě myokardu nejčastěji postiženy, jsou centrální a periferní nervový systém, periferní svalstvo, endokrinní žlázy, dále ledviny, oči, uši, kostní dřeň, střeva, kůže [66]. Typické kombinace klinických manifestací jsou nazývány mitochondriálními syndromy, např. MELAS (z angl. mitochondrial myopathy, encephalopathy, stroke-like episodes), MERRF (z angl. myoclonus epilepsy with ragged red fibres) či syndrom Kearns-Sayre (progresivní externí oftalmoplegie, pigmentová retinitida, mozečková ataxie) [67]. Nezřídka však fenotypické vyjádření mitochondriopatie neodpovídá definici některého ze syndromů a pak hovoříme o tzv. nesyndromickém mitochondriálním onemocnění. V práci Anana et al bylo srdeční postižení zjištěno echokardiograficky či v podobě EKG patologie popsáno u devíti ze 17 nemocných s různými mitochondriálními syndromy v průměrném věku 31 let, tj. u 53 %, přičemž u čtyř jedinců se jednalo o přítomnost kardiomyopatie [68]. Studie provedené na dětských souborech ukazují na prevalenci kardiomyopatického postižení u mitochondriopatií kolem 20–25 % [69,70].
Postižení srdečního svalu se při mitochondriopatiích manifestuje jako hypertrofická (typicky např. u MELAS syndromu) či dilatační kardiomyopatie a je spojeno s horší prognózou, kdy 70 % postižených jedinců umírá do 30 let věku [70]. Hypertrofie LK je nejčastěji koncentrická, může být ale i asymetrická (obr. 6). Stupeň hypertrofie je velmi rozlišný, od mírné až po extrémní. Obstrukce výtokového traktu nebyla dosud u mitochondriální hypertrofické kardiomyopatie popsána. Globální systolická funkce LK je v časných fázích zachována, u většiny nemocných ale dochází k postupnému zhoršování ejekční frakce, jež může být doprovázeno dilatací komory a ztenčováním původně zbytnělých stěn [70]. Rychlost progrese systolické dysfunkce levé komory může být i velmi rychlá, s vývojem terminálního srdečního selhání v řádu několika měsíců. Hypertrofie postihuje často i volnou stěnu pravé komory. Typickými EKG abnormalitami jsou poruchy převodního systému. Lze se setkat s obrazem preexcitace, tj. krátkého PQ intervalu (obr. 7), literárně jsou ale jako nejčastější uváděny různé formy atrioventrikulárních a raménkových blokád [68]. Typicky se tyto vyskytují u syndromu Kearns-Sayre, kde představují jednu ze základních charakteristik tohoto syndromu. Voltážová kritéria hypertrofie LK jsou na EKG obvykle vyjádřena, v individuálních případech ale mohou chybět (obr. 7) [70]. Abnormální myokard je náchylný i ke komorovým arytmiím a náhlá arytmická smrt může být první manifestací mitochondriopatie [71].
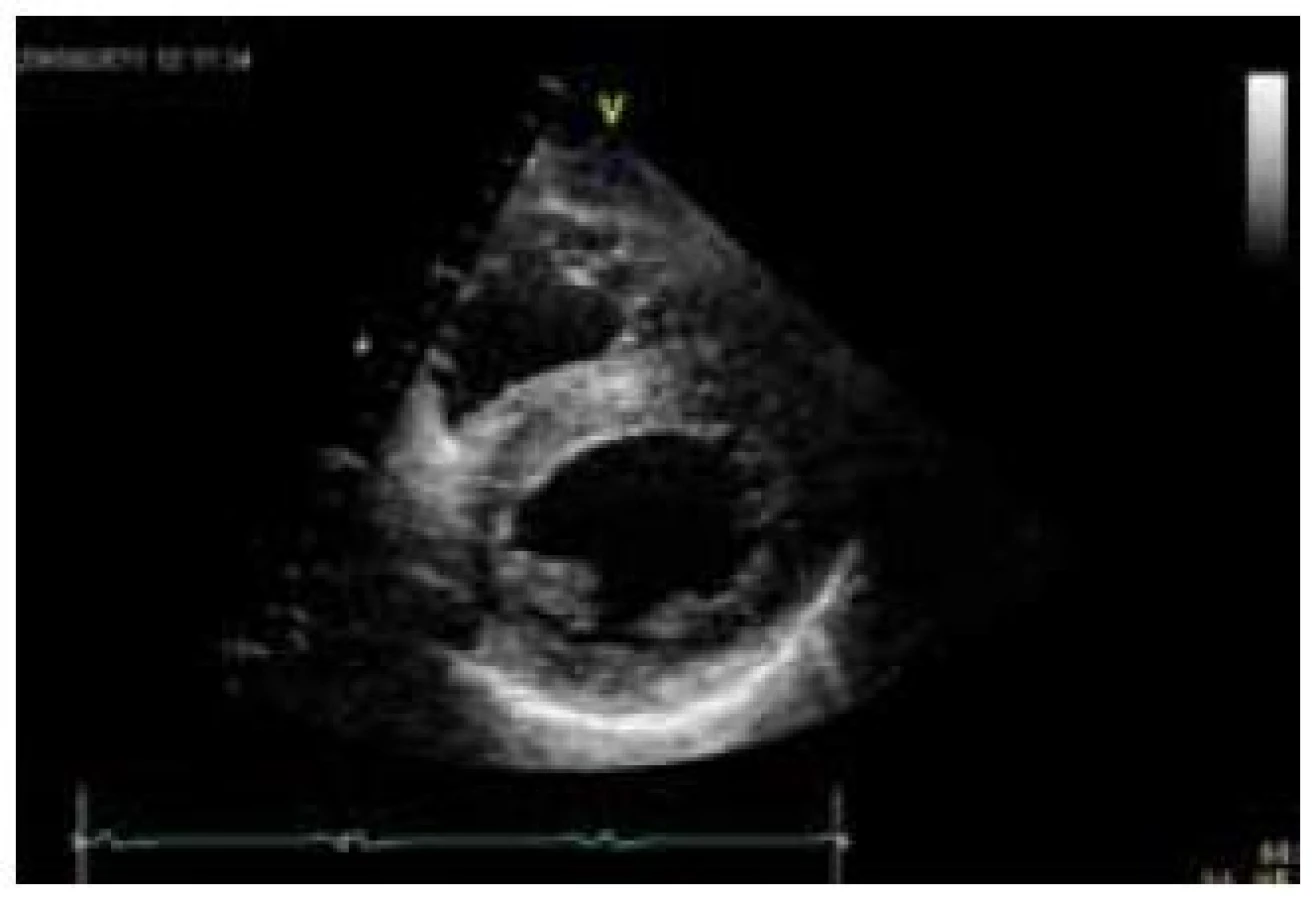
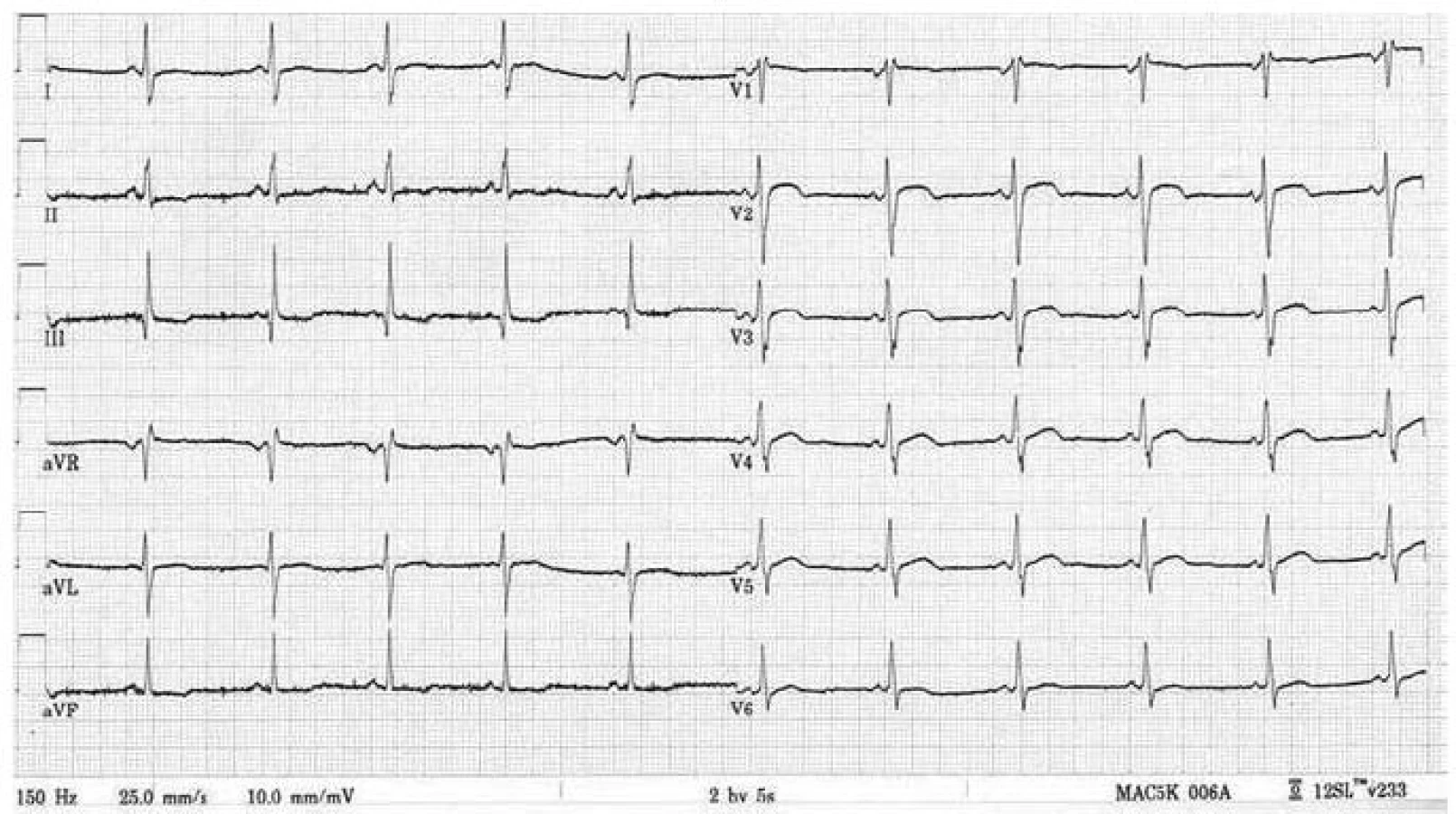
Diagnostika mitochondriálních kardiomyopatií je obtížná. K odhalení myokardiální patologie většinou dochází při kardiologickém screeningu nemocných, kteří se primárně manifestují nekardiální symptomatikou, typicky neuromuskulární. U izolovaného srdečního postižení vede k podezření na mitochondriopatii pečlivá anamnéza, v níž pátráme po, byť i minimálních, projevech postižení jiných orgánů a tkání, především periferního svalstva a centrálního a periferního nervového systému. Velmi důležitým indikátorem jsou zvýšená hladina laktátu v krvi, ať již klidově či excesivně po fyzické zátěži, a zvýšený poměr laktát/pyruvát [65]. Zvýšena může být i aktivita kreatinkinázy. Bioptické vyšetření se primárně provádí z kosterního svalstva; pokud je toto negativní a trvá silné podezření na mitochondriální kardiomyopatii, indikujeme provedení endomyokardiální biopsie [66]. V bioptických vzorcích prokazuje elektronmikroskopické a histochemické vyšetření zvýšený počet a uniformní nebo mozaikovité mitochondriální změny. V kosterních svalech u heteroplazmických mitochondriálních poruch jde o tzv. „ragged red fibres“ (RRF), podmíněných agregací defektních mitochondrií subsarkolemálně. Biochemicky a histochemicky je detekovatelná snížená aktivita a/nebo množství defektního enzymu respiračního řetězce, a to buď uniformě (u nukleárních defektů), nebo mozaikovitě (u mutací mtDNA) [65]. Definitivně je diagnóza mitochondriopatie stanovena molekulárně-genetickým vyšetřením. V případě mitochondriálního onemocnění s nukleárně kódovanou poruchou stačí mutační analýzu provádět v DNA z izolované krve. Jestliže se jedná o mitochondriální onemocnění s maternálním přenosem, tj. mutaci v mtDNA, je s ohledem na možnou nízkou hladinu heteroplazmie hledané mutace v krvi většinou nezbytné provádět mutační analýzu z nejvíce postižené tkáně či z více tkání najednou. Zlatým standardem je vyšetření mtDNA izolované ze svalové tkáně a kultivovaných fibroblastů, ale většinou je vyšetřena i mtDNA izolovaná z buněk močového sedimentu a bukálního stěru. Komplexní laboratorní a genetickou diagnostikou mitochondriopatií se v České republice zabývá Klinika dětského a dorostového lékařství 1. LF UK a Všeobecné fakultní nemocnice v Praze.
Terapie mitochondriálních kardiomyopatií zůstává symptomatickou. Nemocným s mitochondriopatiemi jsou obecně podávány nespecifické dietní doplňky, např. koenzym Q10 a L-karnitin, jejich efekt však nebyl nikdy jasně prokázán ve větších klinických studiích [62]. Při prokázaném kardiálním postižení je nutné pravidelné a relativně časté klinické sledování cílené na monitoraci kvality systolické funkce levé komory, přítomnost symptomů a známek srdečního selhávání, vývoj převodních poruch na EKG a jejich příznaky. Při známkách poklesu systolické funkce LK, resp. manifestaci srdečního selhávání podáváme obvyklou léčbu srdečního selhávání, terapie betablokátory však musí být velmi opatrná vzhledem k možnosti potenciace převodní poruchy. Progredující atrioventrikulární blokády vyžadují implantaci trvalého kardiostimulátoru. Antikoagulační léčba se řídí obvyklými pravidly. Důležité je vyhýbat se lékům, které interferují s funkcí mitochondrií či je přímo toxicky poškozují, např. kortikosteroidy, statiny, fibráty, biguanidy, tetracyklinová antibiotika, doxorubicin [67]. U nemocných s těžkou systolickou dysfunkcí LK a pokročilým srdečním selháním je indikována transplantace srdce [72].
Friedreichova ataxie
Friedreichova ataxie (FA) představuje nejčastější hereditární ataxii. Jedná se o autozomálně recesivní neurodegenerativní onemocnění s frekvencí výskytu 1 : 50 000 v evropské populaci [73]. U více než 98 % postižených jedinců je genetickým podkladem homozygotní mutace spočívající v amplifikaci trinukleotidové repetice guanin-adenin--adenin uvnitř 1. intronu genu X25 na devátém chromozomu, kódujícího strukturu malého mitochondriálního proteinu frataxinu [74]. Velikost expanze výše uvedené repetice negativně koreluje s výsledným fenotypovým projevem onemocnění (objevení se symptomů, jejich tíže, časnost úmrtí) [75]. Zbytek nemocných tvoří jedinci s bodovými mutacemi, což jsou v podstatě složení heterozygoti s expanzí trinukleotidové repetice na jedné alele a bodovou mutací na alele druhé [73]. Frataxin je lokalizován na vnitřní mitochondriální membráně. Předpokládá se, že hraje důležitou úlohu při asemblaci mitochondriálních Fe/S proteinů. Jeho nedostatek má za následek nadměrné mitochondriální hromadění železa a deficit aktivity mitochondriálních Fe/S proteinů, které se uplatňují v respiračním řetězci, což vede ke zvýšenému oxidačnímu stresu [76,77].
K první manifestaci symptomů dochází nejčastěji do 25 let věku, rozmezí je však široké, od dvou do 51 let [78]. Typickými iniciálními příznaky jsou nestabilita při chůzi a nemotornost, které jsou odrazem progredující ataxie [73]. V průměru po 10–15 letech dochází ke ztrátě schopnosti chůze a nutnosti používat invalidní vozík. U většiny postižených je přítomna též dysartrie, pallanestezie a ztráta polohocitu na dolních končetinách, skolióza. Dalšími příznaky mohou být nystagmus, optická atrofie, hluchota, pes cavus a diabetes mellitus [73].
Kardiální postižení se vyskytuje téměř u všech nemocných s FA. K jeho manifestaci dochází obvykle 4–5 let po objevení se neurologických obtíží, v některých případech může být srdeční postižení prvním příznakem onemocnění [79]. Srdeční selhání je zodpovědné za více než 50 % úmrtí nemocných s FA [80]. Histopatologické vyšetření myokardu prokazuje u FA buněčnou hypertrofii, difuzní fibrózu a fokální myokardiální nekrózu, které jsou odrazem dysfunkce mitochondriálních respiračních řetězců [81]. Hypertrofie LK je typickou kardiální manifestací FA. Obvykle bývá koncentrická, i když určitý stupeň asymetrické hypertrofie může být též přítomen (obr. 8) [82]. Tíže hypertrofie nebývá velká, síla stěn zpravidla nepřesahuje 15 mm [80]. Podobně jako u amyloidózy je některými autory u kardiomyopatie při FA popisována granulární textura myokardu [80]. Celková systolická funkce LK je většinou zachována, pouze u některých nemocných je v terminálních fázích srdečního postižení patrna progredující hypokineze stěn a deteriorace ejekční frakce LK, což posléze vede, společně s dilatací a ztenčováním původně hypertrofických stěn komory, k morfologicko-funkčnímu obrazu dilatační kardiomyopatie [46]. I při zachovalé systolické funkci LK lze ale metodami deformační analýzy prokázat lehký stupeň longitudinální kontrakční dysfunkce stěn LK, který koreluje s tíží hypertrofie [83]. Je zajímavé, že některými autory je u většiny nemocných s kardiomyopatií při FA popisována normální diastolická funkce, s čímž koresponduje i absence dilatace levé síně [84,85]. Zesílení volných stěn pravé komory nebývá obvykle vyjádřeno. Vyšetření magnetickou rezonancí prokazuje u přibližně poloviny nemocných midmyokardiální ložiska pozdního sycení kontrastní látkou v oblasti interventrikulárního septa [86]. Nadměrná akumulace železa však není pomocí magnetické rezonance, na rozdíl např. od hemochromatózy, detekovatelná, což je zřejmě podmíněno relativní izolací intramitochondriálních partikulí železa [86].
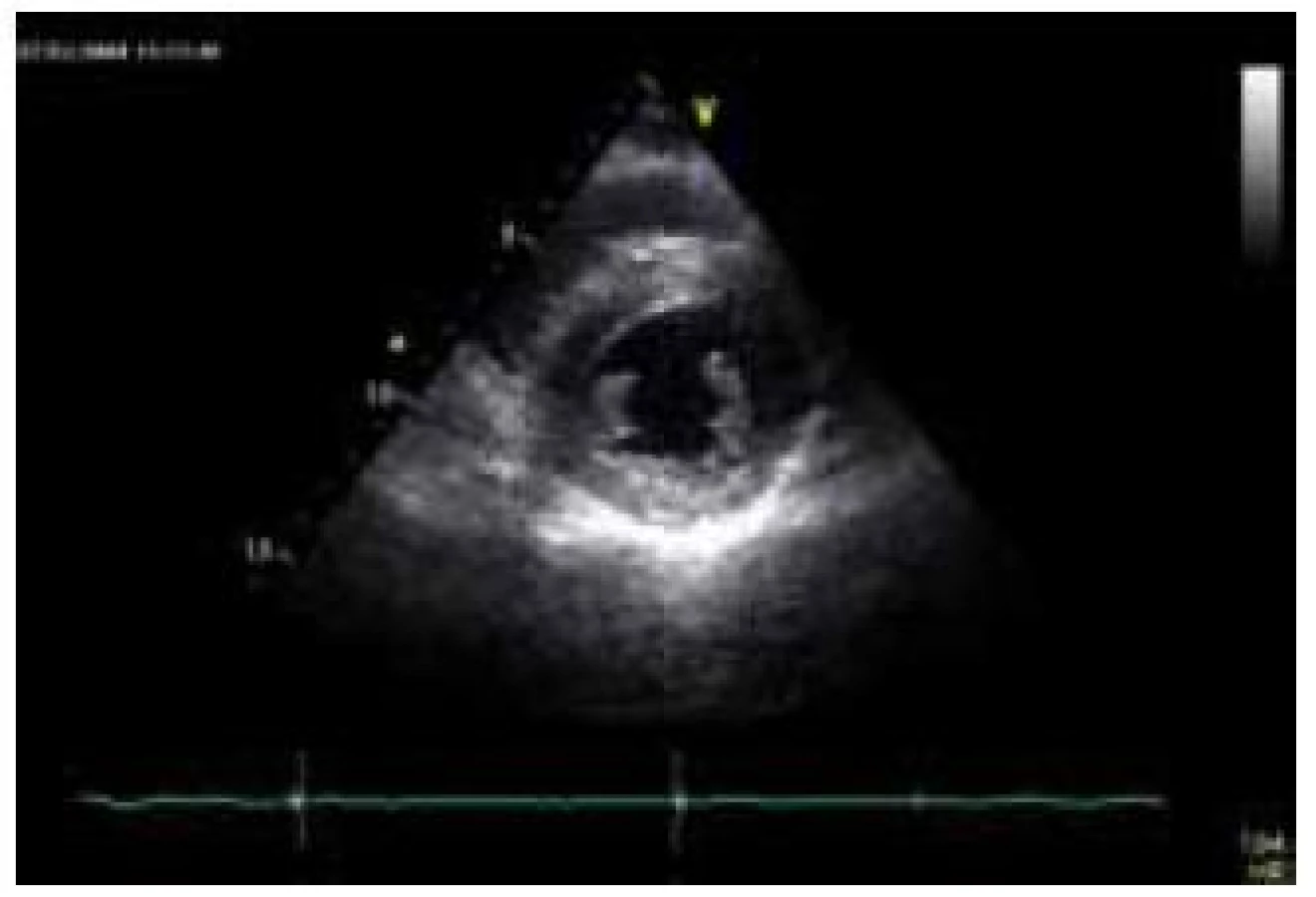
Nejčastější EKG abnormalitou, nalézanou u nemocných s FA, jsou inverze vln T v prekordiálních svodech [79]. Důležitým faktem je, že voltážové známky hypertrofie LK nejsou prakticky nikdy vyjádřeny [46]. Podkladem absence vyšší voltáže komplexu QRS je patrně výrazná přítomnost difuzní fibrózy [87].
Diagnóza FA je založena na přítomnosti typických neurodegenerativních příznaků a molekulárně-genetické analýze [74]. Terapie FA je podpůrná, známky srdečního selhání či arytmické manifestace jsou léčeny konvenčními metodami. V několika studiích byl zkoušen efekt idebenonu, analogu koenzymu Q10 s antioxidačními účinky, na ovlivnění tíže hypertrofie LK, výsledky těchto prací však nepřinesly konzistentní výsledky [88,89].
Ostatní nesarkomerické hypertrofické kardiomyopatie u dospělých
LIM protein. Hlavní úloha svalového LIM proteinu spočívá v podpoře myogeneze a předpokládá se, že působí jako kofaktor při regulaci genové exprese v kosterním svalstvu a myokardu [90]. Vazbou na vlákna aktinu také pravděpodobně stabilizuje propojení kontraktilního aparátu se sarkolemou [91]. Mutace v genu CSRP3, kódujícím strukturu LIM proteinu, byly popsány u jedinců s fenotypickým obrazem hypertrofické i dilatační kardiomyopatie [92]. Přenos je autozomálně dominantně dominantní, s variabilní penetrací.
Myokardiální oxalóza. Primární hyperoxalurie je autozomálně recesivně dědičným onemocněním, při kterém dochází k depozici oxalátových krystalů v různých orgánech, především v srdci a ledvinách [46]. V biopticky odebraných tkáních je prokazatelná depozice dvojlomých krystalků oxalátu, které jsou masivně zmnoženy i v moči. Symetrické zesílení stěn postihuje obě srdeční komory. Celková systolická funkce LK je zpravidla zachována, těžce je však postiženo její plnění [93]. Textura myokardu vykazuje při echokardiografickém vyšetření zřetelně zrnitý vzhled, patrný především na papilárních svalech [94]. U většiny nemocných je na EKG přítomna porucha převodního systému, často v podobě pokročilé atrioventrikulární blokády [46]. Voltáž QRS komplexu může být zvýšená i normální [94].
Mukopolysacharidózy. Mukopolysacharidózy jsou charakterizovány nekompletní lyzozomální degradací kyselých mukopolysacharidů, glykosaminoglykanů [95]. Je logické, že v oblasti srdce postihují zejména mezenchym chlopní a endokard. Dědičnost je autozomálně recesivní, s jedinou výjimkou mukopolysacharidózy typu II, která je vázaná na chromozom X. Určitý stupeň srdečního postižení lze nalézt prakticky u všech typů mukopolysacharidóz, nejtěžší je však přítomno u syndromu Hurlerové, mukopolysacharidózy typu I [46]. Tento syndrom je charakterizován mentální retardací, hepatosplenomegalií, mikrognatií, makroglosií a degenerací sítnice [95]. Hypertrofie LK je často asymetrická, postihující septum komor, se zachovalou ejekční frakcí. Hlavní srdeční abnormalitou je výrazné ztluštění cípů chlopní, především mitrální a aortální, které může vést k hemodynamicky významným vadám, stenotickým i regurgitačním [96]. Na EKG typicky absentují voltážové známky hypertrofie LK [96]. Diagnostika mukopolysacharidóz je prováděna v Ústavu dědičných metabolických poruch 1. LF UK v Praze.
Glykogenózy. S postižením myokardu v podobě hypertrofické kardiomyopatie se lze v dospělosti setkat u glykogenózy typu III (Coriho-Forbesovy nemoci). Jedná se o autozomálně recesivně dědičnou chorobu, podmíněnou deficitem glykogen-odvětvujícího enzymu, amylo-1,6-glukozidázy [97]. Typ IIIa je charakterizován postižením jater, kosterního svalstva a myokardu. Akumulace abnormálního glykogenu vede k obrazu koncentrické hypertrofie spíše malodutinové LK. Echokardiograficky může být dle literatury patrna zrnitá textura myokardu. U některých jedinců dochází ve 3. až 4. dekádě života ke zhoršování původně normální systolické funkce LK a vzniká obraz dilatované hypertrofické kardiomyopatie s nízkou ejekční frakcí [98,99].
Pro úplnost je nutno zmínit, že hypertrofická kardiomyopatie je typickou manifestací nejběžnější metabolické myopatie, glykogenózy typu II (Pompeho choroby). Toto onemocnění je dáno autozomálně recesivně vázanou deficiencí lyzozomální α-glukosidázy [100]. Infantilní forma Pompeho choroby se projevuje již v časném kojeneckém věku a rychle progreduje. Kromě postižení srdečního svalu dominují klinickému obrazu známky svalové hypotonie při postižení skeletálního svalstva, hepatomegalie a makroglosie [98]. Zesílení stěn myokardu srdečních komor je těžké, symetrické. Na EKG je typická velmi vysoká voltáž komplexu QRS, často je zkrácen interval PQ [99]. Diagnostika glykogenóz se v České republice provádí také v Ústavu dědičných metabolických poruch 1. LF UK v Praze.
Kardiomyopatie u poruch oxidace mastných kyselin. Mastné kyseliny představují hlavní zdroj energie pro srdeční sval. Deficity beta-oxidace postihují proces vstupu mastných kyselin do mitochondrie a následnou kaskádu jejich dehydrogenace až do tvorby finálních ketolátek. Tato onemocnění se téměř výhradně manifestují v časném dětském věku. V dospělosti se lze setkat s hypertrofickou kardiomyopatií u deficitu hydroxyacyl-CoA dehydrogenázy mastných kyselin s dlouhým řetězcem (deficit LCHAD) a při deficitu karnitinového přenašeče [101]. Podobně jako u mitochondriálních poruch se diagnostikou zabývá Klinika dětského a dorostového lékařství 1. LF UK a Všeobecné fakultní nemocnice v Praze.
Beckwithův-Wiedemannův syndrom. Hypertrofická kardiomyopatie může být velmi vzácnou součástí klinické manifestace Beckwith-Wiedemannova syndromu, poruchy růstu charakterizované především makrozomií, makroglosií, visceromegalií a omfalokélou či umbilikální kýlou [102].
Syndrom Noonanové. Syndrom Noonanové je autozomálně dominantně dědičnou poruchou, charakterizovanou krátkým vzrůstem, abnormálním obličejem, různými vrozenými srdečními abnormalitami a somatickými známkami Turnerova syndromu při normálním karyotypu [103]. Nejčastějšími kardiálními manifestacemi jsou valvulární stenóza plicnice a hypertrofická kardiomyopatie [82].
Syndrom LEOPARD. Syndrom LEOPARD (z angl. lentigines, ECG abnormalities, ocular hypertelorism, pulmonary stenosis, abnormalities of genitalia, retardation of growth, deafness) je také autozomálně dominantně dědičnou poruchou charakterizovanou retardací růstu, dysmorfií obličeje (zahrnující hypertelorizmus, prognatii, trojúhelníkovitý tvar obličeje, ptózu, malformaci ušních boltců aj.), abnormalitami genitálií, kožními lentigines, hluchotou a kardiálními anomáliemi, především stenózou plicnice a hypertrofickou kardiomyopatií [104]. Hypertrofie myokardu je přítomna u 20 % případů, většinou výrazná, koncentrická, s obstrukcí výtokového traktu LK. Na EKG jsou typicky vyjádřeny různé formy převodních poruch [82].
Závěr
Nesarkomerické formy tvoří malou, ale klinicky důležitou součást fenotypického syndromu hypertrofické kardiomyopatie, a to především ve vztahu k prognóze postiženého individua, genetickému poradenství v rámci rodiny a v řadě případů i stran možnosti specifické terapie, např. u FCH. Většina nesarkomerických fenokopií hypetrofické kardiomyopatie vzniká na podkladě geneticky podmíněné poruchy buněčného metabolizmu, a postižení myokardu je tak jednou ze součástí širšího klinického obrazu charakterizovaného pro danou chorobu typickým multiorgánovým postižením. Na možnost nesarkomerické formy hypertrofické kardiomyopatie je nutné myslet především u nemocných s jiným než autozomálně dominantním typem dědičnosti, extrakardiálními symptomy a klinickými příznaky, při kombinaci hypertrofie LK a převodní poruchy, ať již ve smyslu preexcitace či atrioventrikulárních a raménkových blokád, a při progredující systolické dysfunkci hypertrofické LK. Finální diagnostika nesarkomerických kardiomyopatií patří do rukou specializovaného centra, které se komplexně zabývá danou problematikou.
Věnováno památce profesora Milana Elledera.
Práce byla podpořena Výzkumným záměrem MŠM 0021620806 a MZ0VFN2005.
Doručeno do redakce 25. 9. 2011
Přijato po recenzi 3. 11. 2011
doc. MUDr. Tomáš Paleček, Ph.D.1
MUDr. Petr Kuchynka, Ph.D.1
MUDr. Eduard Němeček1
MUDr. Martin Mašek, Ph.D.2
prof. MUDr. Milan Elleder, DrSc.3
doc. MUDr. Tomáš Honzík, Ph.D.4
prof. MUDr. Aleš Linhart, DrSc.1
1 II. Interní klinika – klinika kardiologie a angiologie, 1. LF UK v Praze a VFN v Praze
2 Radiodiagnostická klinika, 1. LF K v Praze a VFN v Praze
3 Ústav dědičných metabolických poruch, 1. LF UK v Praze a VFN v Praze
4 Klinika dětského a dorostového lékařství, 1. LF UK v Praze a VFN v Praze
tpalec@lf1.cuni.cz
Zdroje
1. Elliott P, Andersson B, Arbustini E et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2008; 29: 270–276.
2. Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA 2002; 287: 1308–1320.
3. Elliott P, McKenna WJ. Hypertrophic cardiomyopathy. Lancet 2004; 363: 1881–1891.
4. Richard P, Charron P, Carrier L et al. EUROGENE Heart Failure Project. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations and implications for molecular diagnosis strategy. Circulation 2003; 107: 2227–2232.
5. Elleder M. Subcellular, cellular and organ pathology of Fabry disease. In: Elstein D, Altarescu G, Beck M (eds). Fabry disease. New York: Springer 2010: 39–71.
6. Dobrovolný R, Dvořáková L, Ledvinová J et al. Relationship between X-inactivation and clinical involvement in Fabry heterozygotes. Eleven novel mutations in the alpha-galactosidase A gene in the Czech and Slovak population. J Mol Med 2005; 83: 647–654.
7. MacDermot KD, Holmes A, Miners AH. Anderson-Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet 2001; 38: 769–775.
8. Meikle PJ, Hopwood JJ, Clague AE et al. Prevalence of lysosomal storage disorders. JAMA 1999; 281: 249–254.
9. Schäffer E, Baron K, Widmer U et al. Thirty-four novel mutations of the GLA gene in 121 patients with Fabry disease. Hum Mutat 2005; 25: 412.
10. Ferrans VJ, Hibbs RG, Burda CD. The heart in Fabry’s disease. A histochemical and electron microscopic study. Am J Cardiol 1969; 24: 95–110.
11. Elleder M, Bradová V, Šmíd F et al. Cardiocyte storage and hypertrophy as a sole manifestation of Fabry’s disease. Report on a case simulating hypertrophic non-obstructive cardiomyopathy. Virchows Arch A Pathol Anat Histopathol 1990; 417: 449–455.
12. Linhart A, Paleček T, Bultas J et al. New insights in cardiac structural changes in patients with Fabry’s disease. Am Heart J 2000; 139: 1101–1108.
13. Weidemann F, Breunig F, Beer M et al. Improvement of cardiac function during enzyme replacement therapy in patients with Fabry disease: a prospective strain rate imaging study. Circulation 2003; 108: 1299–1301.
14. Shah JS, Lee P, Hughes D et al. The natural history of left ventricular systolic function in Anderson--Fabry disease. Heart 2005; 91: 533–534.
15. Paleček T, Dostálová G, Kuchynka P et al. Right ventricular involvement in Fabry disease. J Am Soc Echocardiogr 2008; 21: 1265–1268.
16. Mehta A, Ricci R, Widmer U et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Incest 2004; 34: 236–242.
17. Linhart A, Elliott PM. The heart in Anderson-Fabry disease and other lysosomal storage disorders. Heart 2007; 93: 528–535.
18. Shah JS, Hughes DA, Sachdev B et al. Prevalence and clinical significance of cardiac arrhythmia in Anderson-Fabry disease. Am J Cardiol 2005; 96: 842–846.
19. Linhart A, Lubanda JC, Paleček T et al. Cardiac manifestations in Fabry disease. J Inherit Metab Dis 2001; 24 (Suppl 2): 75–83.
20. Aerts JM, Groener JE, Kuiper S et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc Natl Acad Sci U S A 2008; 105: 2812–2817.
21. Paleček T, Bultas J, Hájek M et al. Association between cardiac energy metabolism and gain of left ventricular mass in Fabry disease. Int J Cardiol 2010; 144: 337–339.
22. Moon JC, Sheppard M, Reed E et al. The histological basis of late gadolinium enhancement cardiovascular magnetic resonance in a patient with Anderson-Fabry disease. J Cardiovasc Magn Reson 2006; 8: 479–482.
23. Moon JC, Sachdev B, Elkington AG et al. Gadolinium enhanced cardiovascular magnetic resonance in Anderson-Fabry disease. Evidence for a disease specific abnormality of the myocardial interstitium. Eur Heart J 2003; 24: 2151–2155.
24. Kawano M, Takenaka T, Otsuji Y et al. Significance of asymmetric basal posterior wall thinning in patients with cardiac Fabry’s disease. Am J Cardiol 2007; 99: 261–263.
25. Clarke JT. Narrative review: Fabry disease. Ann Intern Med 2007; 146: 425–433.
26. Chamoles NA, Blanco M, Gaggioli D. Fabry disease: enzymatic diagnosis in dried blood spots on filter paper. Clin Chim Acta 2001; 308: 195–196.
27. Nakao S, Takenaka T, Maeda M et al. An atypical variant of Fabry’s disease in men with left ventricular hypertrophy. N Engl J Med 1995; 333: 288–293.
28. Sachdev B, Takenaka T, Teraguchi H et al. Prevalence of Anderson-Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation 2002; 105: 1407–1411.
29. Chimenti C, Pieroni M, Morgante E et al. Prevalence of Fabry disease in female patients with late-onset hypertrophic cardiomyopathy. Circulation 2004; 110: 1047–1053.
30. Arad M, Maron BJ, Gorham JM et al. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N Engl J Med 2005; 352: 362–372.
31. Monserrat L, Gimeno-Blanes JR, Marin F et al. Prevalence of Fabry disease in a cohort of 508 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2007; 50: 2399–2403.
32. Paleček T, Linhart A, Magage S et al. Multicentrický screening Fabryho choroby u jedinců s nevysvětlitelnou hypertrofií levé komory. FACSS-desing studie. Cor et Vasa 2005; 47: 242–245.
33. Paleček T, Magage S, Goláň L et al. Kardiální varianta Fabryho choroby manifestující se jako hypertrofická kardiomyopatie s těžkou mid-ventrikulární obstrukcí. Cor Vasa 2009; 51: 11–12.
34. Weidemann F, Linhart A, Monserrat L et al. Cardiac challenges in patients with Fabry disease. Int J Cardiol 2010; 141: 3–10.
35. Keslová-Veselíková J, Hůlková H, Dobrovolný R et al. Replacement of alpha-galactosidase A in Fabry disease: effect on fibroblast cultures compared with biopsied tissues of treated patients. Virchows Arch 2008; 452: 651–665.
36. Koskenvuo JW, Hartiala JJ, Nuutila et al. Twenty-four-month alpha-galactosidase A replacement therapy in Fabry disease has onlyminimal effects on symptoms and cardiovascular parameters. J Inherit Metab Dis 2008; 31: 432–441.
37. Weidemann F, Niemann M, Breunig F et al. Long-term effects of enzyme replacement therapy on Fabry cardiomyopathy: evidence for a better outcome with early treatment. Circulation 2009; 119: 524–529.
38. Pieroni M, Chimenti C, Ricci R et al. Early detection of Fabry cardiomyopathy by tissue Doppler imaging. Circulation 2003; 107: 1978–1984.
39. Weidemann F, Niemann M, Herrmann S et al. A new echocardiographic approach for the detection of non-ischaemic fibrosis in hypertrophic myocardium. Eur Heart J 2007; 28: 3020–3026.
40. Magage S, Linhart A, Bultas J et al. Fabry disease: percutaneous transluminal septal myocardial ablation markedly improved symptomatic left ventricular hypertrophy and outflow tract obstruction in a classically affected male. Echocardiography 2005; 22: 333–339.
41. Danon MJ, Oh SJ, DiMauro S et al. Lysosomal glycogen storage disease with normal acid maltase. Neurology 1981; 31: 51–57.
42. Fukuda M. Biogenesis of the lysosomal membrane. Subcell Biochem 1994; 22: 199–230.
43. Sugie K, Yamamoto A, Murayama K et al. Clinicopathological features of genetically confirmed Danon disease. Neurology 2002; 58: 1773–1778.
44. Boucek D, Jirikowic J, Taylor M. Natural history of Danon disease. Genet Med 2011; 13: 563–568.
45. Charron P, Villard E, Sébillon P et al. Danon’s disease as a cause of hypertrophic cardiomyopathy: a systemic survey. Heart 2004; 90: 842–846.
46. Seward JB, Casaclang-Verzosa G. Infiltrative cardiovascular diseases: cardiomyopathies that look alike. J Am Coll Cardiol 2010; 55: 1769–1779.
47. Maron BJ, Roberts WC, Arad M et al. Clinical outcome and phenotypic expression in LAMP2 cardiomyopathy. JAMA 2009; 301: 1253–1259.
48. Arad M, Moskowitz IP, Patel VV et al. Transgenic mice overexpressing mutant PRKAG2 define the cause of Wolff-Parkinson-White syndrome in glycogen storage cardiomyopathy. Circulation 2003; 107: 2850–2856.
49. Piotrowska-Kownacka D, Kownacki L, Kuch M et al. Cardiovascular magnetic resonance findings in a case of Danon disease. J Cardiovasc Magn Reson 2009; 11: 12.
50. Fanin M, Nascimbeni AC, Fulizio L et al. Generalized lysosome-associated membrane protein-2 defect explains multisystemclinical involvement and allows leukocyte diagnostic screening in Danon disease. Am J Pathol 2006; 168: 1309–1320.
51. Yang Z, McMahon CJ, Smith LR et al. Danon disease as an underrecognized cause of hypertrophic cardiomyopathy in children. Circulation 2005; 112: 1612–1617.
52. Kubánek M, Elleder M, Sikora J, et al. Danonova nemoc – porucha autofagie jako příčina hypertrofické kardiomyopatie. Cor Vasa 2010; 52: 706–712.
53. Echaniz-Laguna A, Mohr M, Epailly E et al. Novel Lamp-2 gene mutation and successful treatment with heart transplantation in a large family with Danon disease. Muscle Nerve 2006; 33: 393–397.
54. Arad M, Benson DW, Perez-Atayde AR et al. Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J Clin Invest 2002; 109: 357–362.
55. Carling D, Woods A, Thornton C et al. Molecular characterization of the AMP-activated protein kinase and its role in cellular metabolism (review). Biochem Soc Trans 1997; 25: 1224–1228.
56. Gollob MH, Green MS, Tang AS et al. Identification of a gene responsible for familial Wolff-Parkinson-White syndrome. N Engl J Med 2001; 344: 1823–31.
57. Murphy RT, Mogensen J, McGarry K et al. Adenosine monophosphate-activated protein kinase disease mimicks hypertrophic cardiomyopathy and Wolff-Parkinson-White syndrome: natural history. J Am Coll Cardiol 2005; 45: 922–930.
58. Burwinkel B, Scott JW, Bührer C et al. Fatal congenital heart glycogenosis caused by a recurrent activating R531Q mutation in the gamma 2-subunit of AMP-activated protein kinase (PRKAG2), not by phosphorylase kinase deficiency. Am J Hum Genet 2005; 76: 1034–1049.
59. Patel VV, Arad M, Moskowitz IP et al. Electrophysiologic characterization and postnatal development of ventricular pre-excitation in a mouse model of cardiac hypertrophy and Wolff-Parkinson-White syndrome. J Am Coll Cardiol 2003; 42: 942–951.
60. Gollob MH. Glycogen storage disease as a unifying mechanism of disease in the PRKAG2 cardiac syndrome. Biochem Soc Trans 2003; 31: 228–231.
61. Wolf CM, Arad M, Ahmad F et al. Reversibility of PRKAG2 glycogen-storage cardiomyopathy and electrophysiological manifestations. Circulation 2008; 117: 144–154.
62. Schapira AH. Mitochondrial disease. Lancet 2006; 368: 70–82.
63. Fosslien E. Review: Mitochondrial medicine – cardiomyopathy caused by defective oxidative phosphorylation. Ann Clin Lab Sci 2003; 33: 371–395.
64. Brega A, Narula J, Arbustini E. Functional, structural, and genetic mitochondrial abnormalities in myocardial diseases. J Nucl Cardiol 2001; 8: 89–97.
65. Marin-Garcia J, Goldenthal MJ. Mitochondrial cardiomyopathy: molecular and biochemical analysis. Pediatr Cardiol 1997; 18: 251–260.
66. Schmiedel J, Jackson S, Schäfer J et al. Mitochondrial cytoptahies. J Neurol 2003; 250: 267–277.
67. Finsterer J. Treatment of mitochondrial disorders. Eur J Paediatr Neur 2010; 14: 29–44.
68. Anan R, Nakagawa M, Miyata M et al. Cardiac involvement in mitochondrial disease. A study on 17 patients with documented mitochondrial DNA defects. Circulation 1995; 91: 955–961.
69. Darin N, Oldfors A, Moslemi AR et al. The incidence of mitochondrial encephalomyopathies in childhood: clinical features, morphological biochemical and DNA abnormalities. Ann Neurol 2001; 49: 377–383.
70. Holmgren D, Wåhlander H, Eriksson BO et al. Cardiomyopathy in children with mitochondrial disease; clinical course and cardiological findings. Eur Heart J 2003; 24: 280–288.
71. Constans J, LeHérissier A, Coquet M et al. Ventricular arrhythmia revealing mitochondrial myopathy in a 69-year-old woman. Eur Heart J 1993; 14: 1137–1139.
72. Santorelli FM, Gagliardi MG, Dionisi-Vici C et al. Hypertrophic cardiomyopathy and mtDNA depletion. Successful treatment with heart transplantation. Neuromuscul Disord 2002; 12: 56–59.
73. Santos R, Lefevre S, Sliwa D et al. Friedreich ataxia: molecular mechanisms, redox considerations, and therapeutic opportunities. Antioxid Redox Signal 2010; 13: 651–690.
74. Koeppen AH. Friedreich’s ataxia: pathology, pathogenesis, and molecular genetics. J Neurol Sci 2011; 303: 1–12.
75. Filla A, De Michele G, Cavalcanti F et al. The relationship between trinucleotide (GAA) repeat length and clinical features in Friedreich ataxia. Am J Hum Genet 1996; 59: 54–60.
76. Foury F, Cazzalini O. Deletion of the yeast homologue of the human gene associated with Friedreich’s ataxia elicits iron accumulation in mitochondria. FEBS Lett 1997; 411: 373–377.
77. Wilson RB, Roof DM. Respiratory deficien-cy due to loss of mitochondrial DNA in yeast lacking the frataxin homologue. Nat Genet 1997; 16: 352–357.
78. Dürr A, Cossee M, Agid Y et al. Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N Engl J Med 1996; 335: 1169–1175.
79. Child JS, Perloff JK, Bach PM et al. Cardiac involvement in Friedreich’s ataxia: a clinical study of 75 patients. J Am Coll Cardiol 1986; 7: 1370–1378.
80. Weidemann F, Niemann M, Ertl G et al. The different faces of echocardiographic left ventricular hypertrophy: clues to the etiology. J Am Soc Echocardiogr 2010; 23: 793–801.
81. Hewer R. The heart in Friedreich’s ataxia. Br Heart J 1969; 31: 5–14.
82. Alizad A, Seward JB. Echocardiographic features of genetic diseases: part 1. Cardiomyopathy. J Am Soc Echocardiogr 2000; 13: 73–86.
83. Weidemann F, Eyskens B, Mertens L et al. Quantification of regional right and left ventricular function by ultrasonic strain rate and strain indexes in Friedreich’s ataxia. Am J Cardiol 2003; 91: 622–626.
84. Giunta A, Maione S, Biagini R et al. Noninvasive assessment of systolic and diastolic function in 50 patients with Friedreich’s ataxia. Cardiology 1988; 75: 321–327.
85. Dutka DP, Donnelly JE, Nihoyannopoulos P et al. Marked variation in the cardiomyopathy associated with Friedreich’s ataxia. Heart 1999; 81: 141–147.
86. Raman SV, Phatak K, Hoyle JC et al. Impaired myocardial perfusion reserve and fibrosis in Friedreich ataxia: a mitochondrial cardiomyopathy with metabolic syndrome. Eur Heart J 2011; 32: 561–567.
87. Isnard R, Kalotka H, Dürr A et al. Correlation between left ventricular hypertrophy and GAA trinucleotide repeat length in Friedreich’s ataxia. Circulation 1997; 95: 2247–2279.
88. Hausse AO, Aggoun Y, Bonnet D et al. Idebenone and reduced cardiac hypertrophy in Friedreich’s ataxia. Heart 2002; 87: 346–349.
89. Lagedrost SJ, Sutton MS, Cohen MS et al. Idebenone in Friedreich ataxia cardiomyopathy-results from a 6-month phase III study (IONIA). Am Heart J 2011; 161: 639–645.
90. Arber S, Halder G, Caroni P. Muscle LIM protein, a novel Essentials regulator of myogenesis, promotes myogenic differentiation. Cell 1994; 79: 221–231.
91. Flick MJ, Konieczny SF. The muscle regulatory and structural protein MLP is a cytoskeletal binding partner of betal-spectrin. J Cell Sci 2000; 113: 1553–1564.
92. Geier C, Perrot A, Ozcelik C et al. Mutations in the human muscle LIM protein gene in families with hypertrophic cardiomyopathy. Circulation 2003; 107: 1390–1395.
93. Palka P, Duhig E, Carey L et al. Primary oxalosis with cardiac involvement: echocardiographic features of an unusual form of cardiomyopathy. Circulation 2001; 103: E122–123.
94. Vélez-Roa S, Depierreux M, Nortier J et al. Cardiac oxalosis: a rare cause of diastolic dysfunction. Eur Heart J 2006; 27: 2496.
95. Muenzer J, Wraith JE, Clarke LA. International Consensus Panel on Management and Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics 2009; 123: 19–29.
96. Rigante D, Segni G. Cardiac structural involvement in mucopolysaccharidoses. Cardiology 2002; 98: 18–20.
97. Kishnani PS, Austin SL, Arn P et al. Glycogen storage disease type III diagnosis and management guidelines. Genet Med 2010; 12: 446–463.
98. Hoffmann GF, Nyhan WL, Zschocke J et al. Dědičné metabolické poruchy. Praha: Grada Publishing, Avicenum 2002: 198.
99. Alizad A, Seward JB. Echocardiographic features of genetic diseases: part 2. Storage disease. J Am Soc Echocardiogr 2000; 13: 164–170.
100. Katzin LW, Amato AA. Pompe disease: a review of the current diagnosis and treatment recommendations in the era of enzyme replacement therapy. J Clin Neuromuscul Dis 2008; 9: 421–431.
101. Spiekerkoetter U. Mitochondrial fatty acid oxidation disorders: clinical presentation of long-chain fatty acid oxidation defects before and after newborn screening. J Inherit Metab Dis 2010; 33: 527–532.
102. Choufani S, Shuman C, Weksberg R. Beckwith--Wiedemann syndrome. Am J Med Genet C Semin Med Genet 2010; 154: 343–354.
103. Tartaglia M, Gelb BD, Zenker M. Noonan syndrome and clinically related disorders. Best Pract Res Clin Endocrinol Metab 2011; 25: 161–179.
104. Grochová I, Groch L. Genetika v kardiologii. Část III. Monogenně dědičné syndromy a kardiologická onemocnění. Cor Vasa 2007; 49: 259–269.
Štítky
Dětská kardiologie Interní lékařství Kardiochirurgie KardiologieČlánek vyšel v časopise
Kardiologická revue – Interní medicína

2011 Číslo 4
Nejčtenější v tomto čísle
- Není hypertrofie jako hypertrofie aneb Nezapomínejme na amyloidózu
- Kalcifikace perikardu
- Nesarkomerické formy hypertrofické kardiomyopatie v dospělosti
- Peripartální kardiomyopatie