
Update role Lp (a) při určení kardiovaskulárního rizika a možnosti jeho ovlivnění
Update of the role of Lp(a) in determination of the CV risk and methods of influencing it
Lipids are transported by lipoproteins in the blood system. Lipoprotein (a) is a unique lipoprotein of the human plasma discovered by Professor Kåre Berg in 1963. Lp(a) is composed of apoliprotein (a) and LDL. In comparison with the other lipoprotein particles, the lipoprotein (a) plasma level is rather constant, only slightly affected by endogenous and exogenous factors. Elevated Lp(a) levels are considered to be a risk factor for atherosclerosis and aortic stenosis. The risk level is over 50 mg/ dl. High levels of Lp(a) have been detected in elderly patients, it is possible that this protective effect supports the reparation of tissue and anticancer activity.
Keywords:
Atherosclerosis – therapy – lipoprotein (a)
Autoři:
Zlatohlávek L.
Působiště autorů:
3. interní klinika 1. LF UK a VFN, Praha
Vyšlo v časopise:
Kardiol Rev Int Med 2019, 21(2): 68-69
Souhrn
Kardiovaskulární onemocnění jsou nejčastější příčinou mortality a morbidity. Kromě „klasických“ rizikových faktorů aterosklerózy jsou známy další rizikové faktory aterosklerózy. Mezi ně patří lipoprotein (a) – Lp (a). Za rizikové hodnoty jsou považovány hladiny přes 80. percentil, tj. 50 mg/ dl. Lp (a) se skládá z apoliporoteinu (a) a LDL částice (apolipoprotein B100). Za rizikovou je zodpovědná nejen zvýšená hladina, ale i velikost Lp (a). Pravděpodobně rizikové jsou vysoké hladiny krátkých izoforem, naopak protektivní mohou být vysoké hladiny dlouhých izoforem. Farmakologicky je dnes možno hladiny Lp (a) ovlivnit PCKS9 inhibitory a pravděpodobně novými léky v klinických studiích.
Klíčová slova:
lipoprotein (a) – ateroskleróza – terapie
Úvod
Kardiovaskulární onemocnění (KVO) jsou nejčastější příčinou mortality a morbidity. Jistě nejsou v dnešní době již zpochybnitelné jasné rizikové faktory aterosklerózy, ať neovlivnitelné (pohlaví, věk, genetická výbava, aj.) nebo ovlivnitelné (dyslipidemie, kouření, nízká pohybová aktivita, obezita, diabetes mellitus, arteriální hypertenze aj.). Nicméně u některých pacientů se stejnými rizikovými faktory je manifestace aterosklerózy různorodá (časností manifestace, rozsahem a místem postižení, aj.). Proto jsou hledány stále nové rizikové faktory, které akcelerují projevy aterosklerózy. Jedním z potencionálních kandidátů je lipoprotein (a) – Lp (a), který se po objevení v 70. letech dostává zpět do světla vědeckého
zájmu.
Lp (a) je plazmatický lipoprotein tvořený z apoliporoteinu (a) – apo (a) a LDL částice (apolipoprotein B100 – apoB-100). Dle klinických studií a studií na zvířecích modelech jsou zvýšené hladiny Lp (a) spojené se zvýšeným rizikem aterosklerózy a stenózy aortální chlopně. Za rizikové hodnoty jsou považovány hladiny přes 35 mg/ dl, resp. 80. percentil, tj. 50 mg/ dl. Princip jakým se Lp (a) uplatňuje v procesu aterogeneze není zcela znám. Roli hraje pravděpodobně jeho funkce při reparaci a hojení ran, kdy Lp (a) „přináší“ do poškozeného endotelu lipidy jako substrátu k hojení, čímž paradoxně přispívá k ukládání dalších lipidů do ateroslerotického plátu. Další jeho jistě významnou rolí je vysoká homologie s plazminogenem. Jeho vazbou na receptory pro plazminogen inhibuje jako afunkční molekula fibrinolýzu a tím napomáhá
trombogenezi.
Naopak překvapivě vysoké hladiny Lp (a) ve stáří jsou asociovány s dlouhověkostí. Vysvětlení tohoto faktu je možno hledat právě v jeho roli při reparaci tkání, hojení ran a protinádorovém působení.
Hladina a velikost Lp (a) jsou u člověka velmi variabilní a jsou dány množstvím syntetizovaného apo (a). Gen pro Lp (a) je lokalizován na 6. chromozómu. Hladina Lp (a) je z více jak 95 % dány geneticky a poměrně stabilní po celou dobu života., Mohou ovlivňovat následující faktory – pohlaví, některé hormony a léky. Hladina není prakticky ovlivnitelná dietou.
Lp (a) je přítomen u člověka, primátů a ježka evropského, jeho fyziologické a patofyziologické funkce nebyly stále jasně objasněny. Lp (a) je jeden z dalších potenciálních kandidátů ať aditivního či samostatného rizikového faktoru aterosklerózy.
Lp (a) – lipoprotein (a); apo (a) – apolipoprotein
(a); apoB-100 – apolipoprotein
B-100, LDL – lipoprotein s nízkou hustotou
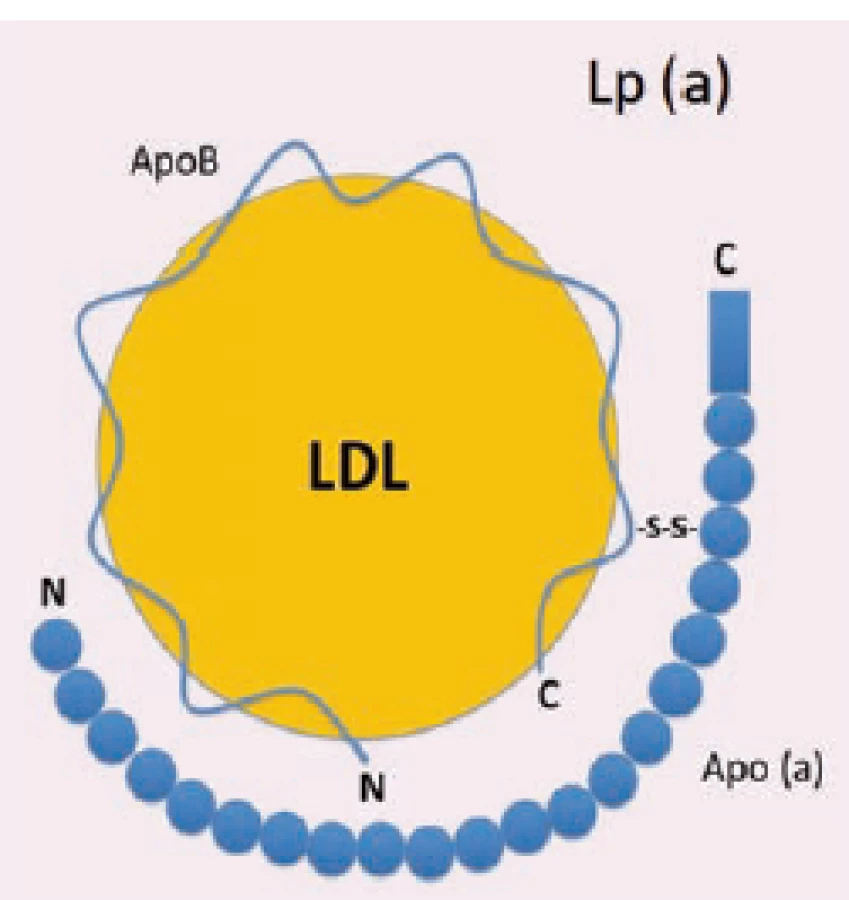
Lipoprotein (a)
První, kdo nevědomky objevil Lp (a), byl prof. Berg v roce 1963. Objevil v plazmě antigen, který přiřadil do oblasti lipoproteinů [1]. Původně byl tento antigen spojován s LDL (lipoprotein s nízkou hustotou) částicí, ale Berg ukázal, že tomu tak není, že se jedná o antigen reprezentující odlišnou lipoproteinovou částici a označil ji jako Lp (a) [2]. Po zveřejnění výsledků prof. Berga objevil prof. Seeger [3] pomocí elektroforézy lipoprotein, o kterém se domníval, že se jedná o genetickou variantu
β-migrující LDL. Následně bylo dokázáno, že se jedná o nový lipoprotein pohybující se v oblasti pre β [4]. V roce 1970 popsal prof. Rider lipoprotein, který se sice elektroforeticky choval jako lipoproteiny o velmi nízké hustotě (VLDL), ale ultracentrifugací byl porovnatelný s LDL. Všechny práce popisovaly ve skutečnosti Lp (a). Název Lp „(a)“ vznikl jako označení lipoproteinu s antigenními vlastnostmi.
Lp (a) má zhruba sférický tvar, o průměru 21 nm [5]. Skládá se z "low-density" jádra obaleného "high-density" pláštěm [6]. Stejně jako LDL částice obsahuje apoB-100 a dále druhý, disulfidicky vázaný polypeptid-apolipoprotein (a) [7]. Apo (a) je vysoce glykosylovaný, hydrofilní protein s velmi malou afinitou k lipidům, který vykazuje rozmanitou délkovou variabilitu [8]. Sestává se z domén, tzv. kringlů (kringl = z dánštiny preclík) a serin-proteázové domény. Kringly mají strukturu trojité smyčky, která je stabilizována třemi disulfidickými vazbami.
Kringly se vyskytují také v plazminogenu, protrombinu, tkáňovém aktivátoru plazminogenu a dalších proteázách podílejících se na koagulaci a fibrinolýze. Lp (a) obsahuje 11 variant kringlů [9]. Poslední z nich je z 85 % homologní s kringlem V. plazminogenu. Zbývajících deset je podobných, ale nikoli identických, s kringlem IV. plazminogenu. Těchto deset je označeno jako kringl IV typy 1–10. Kringly IV typ 1 a 3–10 jsou přítomny pouze v jedné kopii, oproti tomu typ 2 se vyskytuje v různých počtech opakování, a to 3–43× [10]. A právě tato repetice kringlu IV typ 2 udává rozdílnou velikost apo (a).
Plazminogen se díky své vlastnosti vázat lysin váže na řadu specifických substrátů [11] (např. fibrin, buněčné receptory, endotelové buňky tepen). Kringl IV typ 10 lidského apo (a) má taktéž schopnost se vázat na stejné substráty jako plazminogen, s nímž interferuje jako nefunkční proteáza, zejména při trombolýze.
Gen pro apo (a) je lokalizován na 6. chromozomu v oblasti 6q26-q27 asi 50 kb od genu pro plazminogen, se kterým sdílí asi 80% homologii. Jeho rozsáhlost je dána mnohonásobným opakováním 5,5 kb dlouhé jednotky kódující kringl IV typ 2 [12,13] objevující se v rozdílném počtu repetic. Exprese genu probíhá v jaterní buňce.
Míra exprese genu je ovlivňována jeho regulačními oblastmi, dominantně v promotorové oblasti a oblasti zesilovačů, kde se pravděpodobně uplatňují farmakologické vlivy a vlivy vnitřní regulace [14].
Metabolizmus
Množství kolujícího Lp (a) je dáno zejména množstvím produkce, nikoliv jeho degradace [15]. Lp (a) je syntetizován dominantně v játrech [16]. Určujícím faktorem hladiny Lp (a) je množství apo (a), jehož hladiny v jaterní buňce jsou individuální dle míry transkripce a stability příslušné mRNA. V endoplazmatickém retikulu každý kringl prochází sérií posttranslačních modifikací vč. formace tří disulfidických vazeb a přidání N-vázaných glykanů. Delší formy apo (a) jsou během transportu z endoplazmatického retikula déle zadržovány a mohou být degradovány proteozomy. Malé formy jsou efektivněji transportovány na místo dalšího zpracování, do Golgiho komplexu, kde dochází k dalšímu navázání na N- a O- vázaných glykanů. Nově syntetizovaný apo (a) je transportován na povrch jaterní buňky, kde je shromažďován s lipoproteiny bohatými na apo-B100. Jak bylo popsáno výše, velké formy jsou během posttranslačních úprav více náchylné k degradaci, a proto jsou spojeny s nižšími sérovými hladinami Lp (a), u malých forem je tomu naopak.
Celkem 10–25 % Lp (a) je konvertováno na LDL částice, které jsou z oběhu odstraňovány LDL-receptorem [17]. Při pokusech na tkáňových kulturách VLDL [18] receptor internalizoval Lp (a). U myší s "knockoutovaným" VLDL receptorem dochází ke zvýšení hladin Lp (a).
Ledviny se také podílí na odstranění Lp (a) z oběhu [19]. Byl zjištěn arterio-venózní gradient v hladinách Lp (a) ledvinných tepen a žil. Pacienti s renální insuficiencí mají zvýšené hladiny Lp (a). Princip jakým velké glykosylované substráty prochází do moče, není přesně znám.
Hladina Lp (a) minimálně ovlivnitelná věkem, pohlavím či dietou a jeho hladina je poměrně stabilní po celou dobu života. U žen po menopauze dochází pravděpodobně vlivem nedostatku estrogenů k vzestupu hladiny Lp (a) o 10–30 %. Estrogeny a kyselina nikotinová působí na úrovni transkripce snížením hladiny příslušné mRNA. Růstový hormon působí opačně.
Fyziologické funkce
Lp (a) je především považován za aterogenní rizikový faktor, nicméně má pravděpodobně i fyziologické funkce. Jedna z možných rolí Lp (a) je jeho úloha při reparaci a hojení ran [17].
Lp (a) vstupuje do procesu hojení po kontaktu extracelulární matrix poraněné tkáně s krevním proudem [20]. LDL část Lp (a) dodává lipidy pro reparaci tkáně. Lp (a) podporuje další influx ostatních zánětlivých buněk, např. má chemotaktický vliv na monocyty. Dle studií in vitro [21] je Lp (a) schopen fungovat jako scavenger akumulovaných a nadbytečných biologických lipidů. Pomocí repetic domény kringl IV je schopen navázat zbytky metabolizmu (vč. oxidovaných lipidů) a nabídnout je k dalšímu zpracování.
Lp (a) je přítomen v blízkosti endoteliálních buněk novotvořených cév, avšak nebyl již detekován v lézích po epiteliální regeneraci [22]. In vitro dochází vlivem Lp (a) k inhibici novotvorby cév v nádorovém procesu pravděpodobně zablokováním receptorů pro angiostatin, kterému je strukturou podobný.
Pozoruhodné jsou studie se stoletými lidmi [23] u nichž byla zjištěna spíše vyšší hladina Lp (a) než u srovnávací skupiny středního věku. Zdá se, že pokud nejsou přítomné jiné rizikové faktory aterosklerózy, Lp (a) se spíše uplatňuje v procesu hojení ran a svými možnými antineoplazmatickými vlastnostmi. Pravděpodobně ochranný, protektivní vliv mají vyšší hladiny středně dlouhých délkových variant Lp (a).
Aterogeneze
Řadou studií a pokusy in vitro bylo prokázáno, že se Lp (a) akumuluje v aterosklerotickém plátu, ale nikoli ve zdravé stěně cév. Většina prací studujících aterogenezi Lp (a) byla prováděna in vitro, kde byly sledované parametry izolované, a tudíž plně neodráží situaci in vivo.
V časné fázi tvorby plaku se Lp (a) nalézá v blízkosti endoteliálních buněk, transport je pravděpodobně zprostředkován VLDL receptory, které jsou přítomny na kapilárách a drobných cévách. Lp (a) je zachycen v lézích vazbou na komponenty extracelulární matrix. Lp (a) zde zvyšuje expresi adhezivních molekul a napomáhá chemotaxi zánětlivých buněk [25]. Výsledkem interference s plazminem dochází k snížení aktivity TGF (transformující růstový faktor) a tím ke zvýšení proliferace buněk hladké svaloviny. Lp (a) je v plaku často lokalizován v blízkosti pěnových buněk, s jejichž zbytky a dalšími látkami (např. oxidovanými lipidy) je "scavengován" do makrofágů [26]. Tím zvyšuje Lp (a) lokální depozita cholesterolu v lézích a napomáhá k rozvoji plaku. Lp (a) se dále akumuluje v blízkosti krevních destiček [27], kompetuje s plazminogenem o vazbu na fibrin a zpomaluje aktivaci tkáňového aktivátoru plazminogenu, inhibuje lokálně trombolýzu [28]. V pokročilých lézích je apo (a) predominantně lokalizován extracelulárně ve ztluštělé intimě. Lp (a) nebyl nalezen ve zdravé intimě cév, ale vždy v aterosklerózou poškozených cévách.
Lp (a) a kardiovaskulární riziko
Brzy po objevení Lp (a) se tento lipoprotein dostal do světla zájmu ve vztahu k procesu aterosklerózy. Byla provedena řada studií, hledajících vztah Lp (a) k ICHS. Vzhledem k rozdílné izolaci, skladování a chemické analýze Lp (a), i k odlišnému designu studií, počtu vyšetřených pacientů a různé statistické analýze jsou výsledky provedených klinických studií obtížně srovnatelné, ale lze říci, že Lp (a) je nezávislý rizikový faktor v rozvoji aterosklerózy, okluzi periferních tepen a stenózy aortální chlopně [29,30].
Podobné výsledky vyšly z retrospektivních a case-control studií (tab. 1). Některá věrohodná data ukazují, že zvýšené hladiny Lp (a) jsou prediktorem progrese preexistujích změn, které byly monitorovány angiograficky.
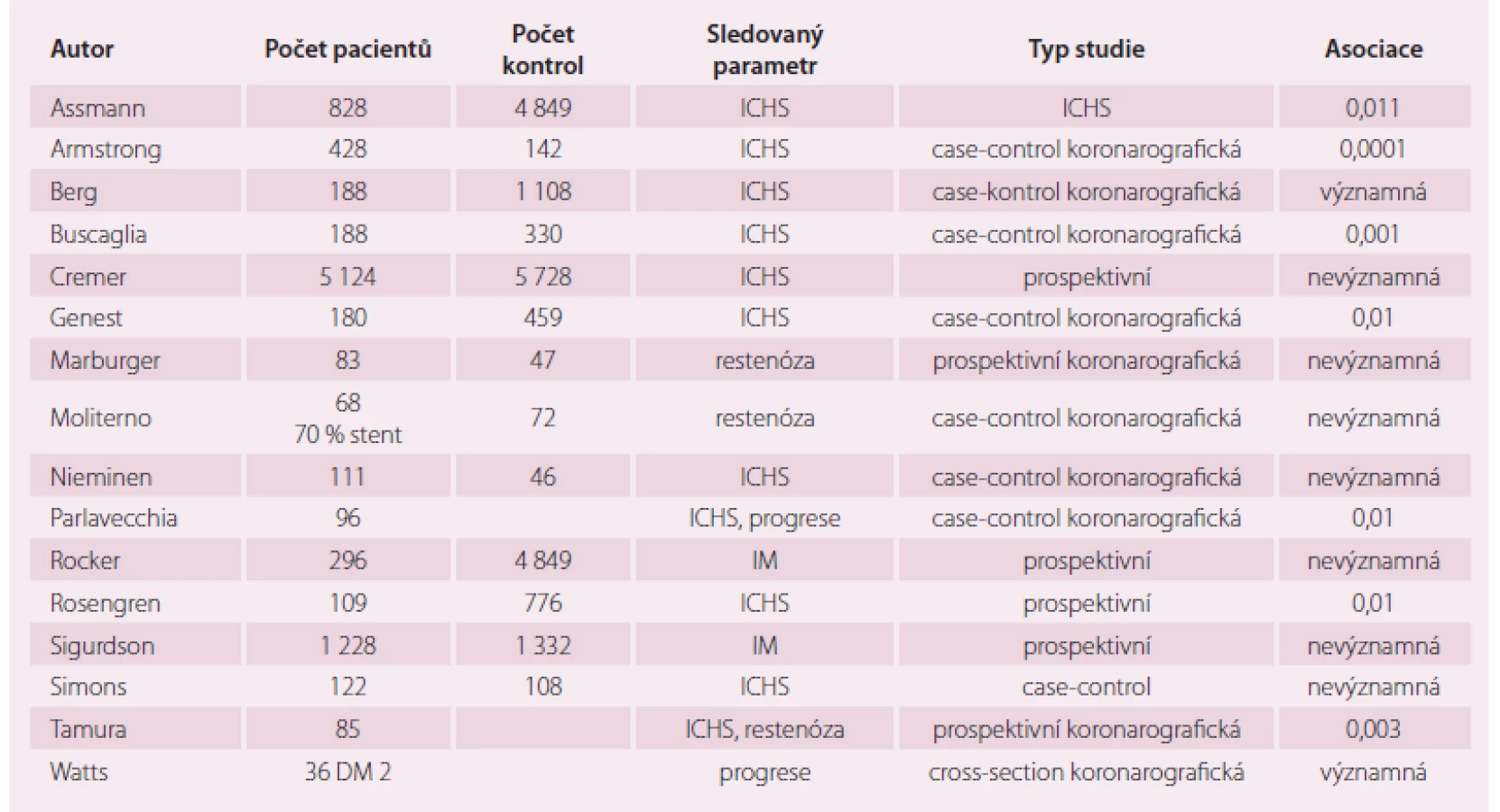
Vztah Lp (a) a cévní mozkové příhody byl studován [31] v 16 epidemiologických studiích, z jejichž závěrů nevyplynul Lp (a) jako prediktor ischemické mozkové příhody, ale může být pomocný rizikový faktor iktu u pacientů s jiným metabolickým postižením. Jisté podezření vztahu embolických příhod a Lp (a) vzhledem k "oslabené" fibrinolýze nelze opominout.
Jistě významnou a velice často v odborné literatuře citovanou rolí Lp (a) je jeho vztah k okluzivním komplikacím po různých intervenčních výkonech jako je: perkutánní transluminální koronarografická angioplastika [32], stentování koronárních tepen, kde se uplatňuje samostatně, či samozřejmě v kombinaci s ostatními rizikovými faktory. Vysvětlením těchto komplikací je zřejmě inkorporace Lp (a) do stěn tepen během novotvoření intimy po poškození zákrokem. Z provedených studií lze říci, že hladina Lp (a) přes 30 mg/ dl je rizikovým faktorem okluze, samozřejmě potencována ostatní rizikovými faktory jako jsou např.: vysoké hladiny LDL, nízké hladiny HDL.
Vzhledem k možnému negativnímu vlivu na fibrinolýzu je diskutován vztah Lp (a) k žilním trombózám [33], uzávěrům retinálních žil a placentární cirkulace jako příčině retardace růstu plodu. Nelze ale jednoznačně říci, že Lp (a) má jasný synegrický efekt spolu s dříve definovanými a známými protrombotickými prediktory.
Hladiny Lp (a) a možnosti jejich ovlivnění
Bylo provedeno mnoho studií zkoumajících vliv diety na hladinu Lp (a). Byl zkoumán vliv vysokocholesterolové diety [34], po které došlo ke zvýšení apo-B100, ale nikoliv Lp (a). Stejně tak nízkokalorická dieta ani rybí olej neměly vliv na hladinu Lp (a). U žen, po redukci váhy o 10 kg došlo k statisticky významnému poklesu Lp (a), ale nebyl pozorován efekt u žen se stejným váhovým úbytkem, které měly hladinu Lp (a) pod 30 mg/ dl.
Kyselina nikotinová byl první objevený lék ovlivňující hladinu Lp (a). U hypercholesterolemických pacientů s hladinou Lp (a) přes 30 mg/ dl došlo k poklesu Lp (a) o 36 %, avšak 45 % pacientů studii pro nežádoucí účinky nedokončilo [35]. Analog kyseliny nikotinové Acipimox v dávce 1g redukuje hladinu Lp (a) o 8 % méně než kyselina nikotinová. Snížení hladiny Lp (a) bude zřejmě způsobeno ovlivněním jeho syntézy na úrovni regulačních oblastí genu pro apo (a). Kyselina nikotinová také snižuje lipolýzu tukové tkáně, a tím dodání mastných kyselin do jater. Následkem je snížená hladina VLDL částic a vzestupu počtu VLDL receptorů, pomocí kterých je Lp (a) pravděpodobně také vychytáván. Tento druhý princip se ovšem na snížení Lp (a) podílí minimálně.
Ve dvojitě slepé, placebem kontrolované studii [36] při podávání gemfibrozilu v dávce 50 mg/ kg/ den došlo ke snížení Lp (a) o 16 %. Po podávání probukolu nebyl pozorován vliv na hladinu Lp (a).
Statiny dle většiny studií neovlivňují hladinu Lp (a) a v několika menších pozorováních došlo dokonce ke zvýšení Lp (a) [37].
Zaručenou metodou snižující hladinu Lp (a) je aferéza [38]. Princip je stejný jako při LDL-aferéze. V preventivní studii sledující restenózu po PTCA (perkutánní transluminární koronární angiografii) bylo sledováno výrazné snížení těchto příhod o více než 50 % po redukci koncentrace Lp (a) aferézou.
Prvním hormonem, se kterým byla provedena studie prokazující pozitivní vliv na Lp (a), byl analog androgenu stanozol, ale i jiné androgeny snižují hladiny Lp (a). U pacientů s karcinomem prostaty dochází po orchiektomii ke zvýšení hladin Lp (a), kdežto u pacientů léčených estrogeny dochází k poklesu Lp (a) [39]. V mnoha studiích postmenopauzálních žen bylo po podávání monoterapie estrogeny či kombinované terapie s progesteronem pozorováno snížení Lp (a). Stejný vliv byl pozorován po podávání antiestrogenu tamoxifenu.
Z ostatních hormonů zvyšuje hladinu Lp (a) např. růstový hormon, naopak dexametazon nebo adenokortikotropní hormon hladinu Lp (a) snižují. Nálezy u thyreoidních hormonů jsou rozporuplné.
Nadějnou lékovou skupinou jsou inhibitory PCSK9. Již v roce 2015 publikovaná metaanalýza 6 566 pacientů potvrdila pokles Lp (a) o 26 % po podávání této skupiny léčiv [40]. Nicméně pokles KV mortality nebyl asociován s poklesem Lp (a) v těchto metaanalýzách.
Ve studii ODYSSEY [41] s alirocumabem došlo k výraznému snížením Lp (a), bez ohledu na počáteční dávku a užívání statinů. Ve 24. týdnu došlo ke snížení Lp (a) oproti výchozím hodnotám o 23–29 % dle síly preparátu v dávce 75 či 150 mg podávaný 1× za 14 dní (všechna srovnání p < 0,0001 oproti kontrolám). Snížení bylo udržováno v průběhu sledování až do 104. týdne. Pokles Lp (a) byl nezávislý na rase, pohlaví, přítomnosti familiární hypercholesterolemie, výchozích koncentracích Lp (a) a LDL cholesterolu nebo použití statinů.
Identicky ve studii s evalocumabem [42] došlo po 48 týdnech k poklesu Lp (a) o 26,9 %. Evolocumab snížil riziko vzniku ICHS, infarktu myokardu nebo urgentní revaskularizace o 23 % u pacientů s výchozím Lp (a) > medián a o 7 % u pacientů s hodnotami ≤ medián. Tedy, evalocumab významně snížil hladiny Lp (a) a pacienti s vyššími hodnotami Lp (a) zaznamenali větší absolutní snížení Lp (a) a inklinovali k většímu koronárnímu prospěchu z inhibice PCSK9.
Další novou nadějí a úspěšnou farmakoterapii má ve svém vývoji firma Ionis Pharmaceuticals, a to preparát IONIS-APO(a)LRx podávaný 1× týdně subkutánně [43]. Jedná se o antisense oligonukleotidy RNA. Ve druhé fázi klinického zkoušení došlo dle dávky k poklesu hladiny Lp (a) od 66 do 92 %, bez významných vedlejších nežádoucích efektů. Tento preparát je nyní v dalších fázích klinického výzkumu. Antisense terapie je tedy nejen potenciálně léčebná metoda aterosklerózy, ale i vysokých hladin Lp (a).
Doporučení konsensu EAS
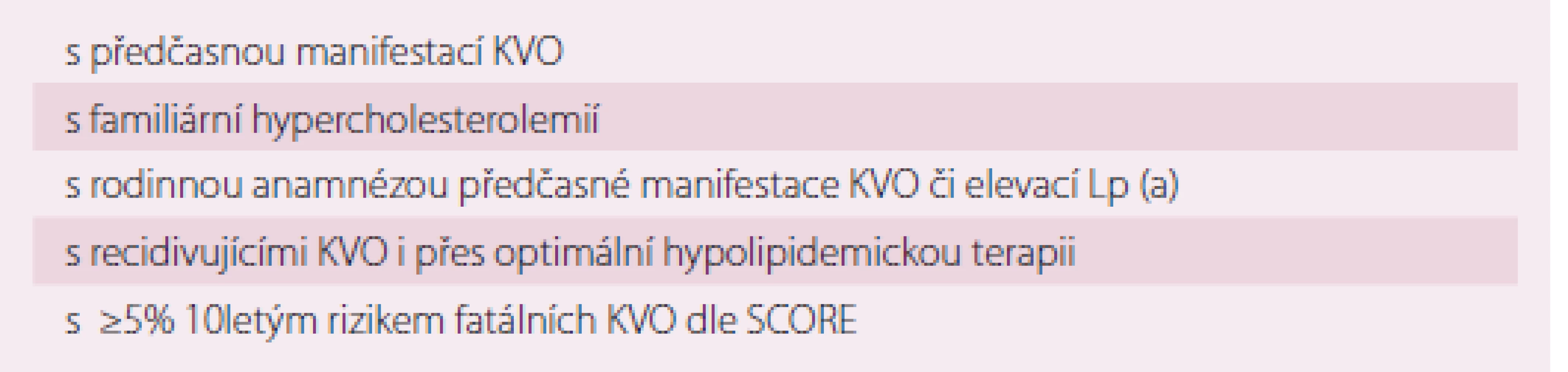
Dle tohoto doporučení by vzhledem k t. č. nemožnosti dostatečného farmakologického ovlivnění neměl být prováděn plošný screening Lp (a), avšak měření Lp (a) by mělo být systematicky zvažováno u osob s vysokým rizikem KVO nebo s pozitivní rodinnou anamnézou předčasné manifestace ICHS či vysokých hladin Lp (a) (tab. 2) [44]. Riziko je považováno za významné, pokud je hladina Lp (a) nad 80. percentilem, tj. nad 50 mg/ dl. U těchto pacientů by mělo dojít k překlasifikování jejich rizika a měli by být považováni za pacienty s vysokým rizikem. Vhodná volba pro pacienty s rizikem a vysokou hladinou Lp (a) je intenzivní léčba rizikových faktorů.
Závěr
Lp (a) je unikátní lipoproteinová částice jednak svou genetickou determinací, ale i strukturou. Lp (a) hraje zřejmě významnou úlohu při hojení poškozených tkání. Naopak svým působením v poškozeném endotelu zhoršuje proces aterogeneze a homologií s plazminogenem inhibuje fibrinolýzu. Na základě studií sledujících asociaci mezi zvýšenými koncentracemi Lp (a) a zvýšeným rizikem ischemické choroby lze Lp (a) považovat za samostatný rizikový faktor aterosklerózy a stenózy aortální chlopně. V léčbě zvýšených hladin Lp (a) máme omezené možnosti. Dietní intervence není úspěšná, z farmakologických prostředků snižuje hladinu Lp (a) významně kyselina nikotinová a její deriváty, které nejsou t. č. k dispozici, statiny jsou bez efektu. Novou nadějí jsou PCSK9 inhibitory a antisense apo(a) RNA terapie.
Doručeno do redakce: 29. 5. 2019
Přijato po recenzi: 6. 6. 2019
doc. MU Dr. Lukáš Zlatohlávek, Ph.D.
www.lf1.cuni.cz
Zdroje
1. Berg K. A new serum type system in man: the Lp sytem. Acta Pathol Microbial Scand 1963; 59: 369–382.
2. Morrisett JD, Guyton JR, Gaubatz JW et al. Lipoprotein (a): structure, metabolism and epidemiology. In: Plasma lipoproteins. Gotto AM. (ed). New Comprehensive Biochemistry. Elsevier Sci Publ 1987; 14: 129–152. doi: 10.1016/ S0167-7306(08)60198-2.
3. Seegers W, Hirschhorn K, Burnett L et al. Double beta-lipoprotein: a new genetic variant in man. Science 1965; 149(3681): 303–304. doi: 10.1126/ science.149.3681.303.
4. Dahlén G, Ericson C, Furberg C et al. Studies on an extra pre-beta lipoprotein fraction. Acta Med Scand Suppl 1972; 531: 1–29.
5. Sines J, Rothnagel R, van Heel M et al. Electron cryomycroscopy and digital image processing of Lp(a). Chem Phys Lipids 1994; 67/ 68: 81–89.
6. Marcovina SM, Morrisett JD. Structure and metabolism of lipoprotein (a). Curr Opin Lipidol 1995; 6(3): 136–145.
7. Koschinsky ML, Cote GP, Gabel B et al. Identification of the cystein residue in apolipoprotein(a) that mediates extracellular couping with apolipoprotein B-100. J Bioch Chem 1993; 268(26): 1819–19825.
8. Hixson JE, Britten ML, Manis GS et al. Apolipoprotein(a) glycoprotein isoform result from size differences in Apo(a) mRNA in baboons. J Biol Chem 1989; 264(11): 6013–6016.
9. Morrisett JD, Gaubatz JW, Knapp RD et al. Structural properties of apo (a): a major apoprotein of human lipoprotein (a). In: Lipoprotein (a). Scanu AM (ed). San Diego: Academic Press Ic. 1990: 53–74.
10. Lackner C, Cohen JC, Hobs HH. Molecular definition of the extreme size polymorphism in apolipoprotein(a). Hum Mol Genet 1993; 2(7): 933–940. doi: 10.1093/ hmg/ 2.7.933.
11. Eaton DL, Fless GM, Kohr W jet al. Partial amino acid sequence of apolipoprotein(a) shows that it is homologous to plasminogen. Proc Natl Acad Sci USA 1987; 84(10): 3224–3228. doi: 10.1073/ pnas.84.10.3224.
12. White AL, Hixson JE, Rainwater DL et al. Molecular basis for „null“ lipoprotein(a) phenotyps and influence of apolipoprotein(a) size on plasma lipoprotein(a) level in the baboon. J Biol Chem 1994; 269(12): 9060–9066.
13. Lackner C, Boerwinkle E, Leffert C et al. Molecular basis of apolipoprotein(a) isoform size heterogenity as revealed by pulsed-field gel electrophoresis. J Clin Invest 1991; 87(6): 2153–2161. doi: 10.1172/ JCI115248.
14. Boerwinkle E. Genetics of plasma lipoprotein (a) concentrations. Curr Opin Lipidology 1992; 3: 128–136.
15. Krempler F, Kostner GM, Bolzano K et al. Turnover of lipoprotein (a) in man. J Clin Invest 1980; 62(6): 1483–1490. doi: 10.1172/ JCI109813.
16. Kraft HG, Menzel HJ, Hoppichler F et al. Changes of genetics apolipoprotein phenotypes caused by liver transplantation. Implication for apolipoprotein synthesis. J Clin Invest 1989; 83(1): 137–142. doi: 10.1172/ JCI113849.
17. Hobbs HH, White AL. Lipoprotein(a): intrigues and insights. Curr Opin Lipidol 1999; 10(3): 225–236.
18. Argraves KM, Kozarsky KF, Fallon JT et al. The atherogenic lipoprotein Lp(a) is internalized and degraded in a process mediated by the VLDL receptor. J Clin Invest 1997; 100(9): 2170–2181. doi: 10.1172/ JCI119753.
19. Parra HJ, Mezdour H, Cachera C et al. Lp(a) lipoprotein in patients with chronical renal failure treated by hemodialysis. Clin Chem 1987; 33(5): 721.
20. Higazi AA, Lavi E, Bdeir K et al. Defensin stimulated the binding of lipoprotein (a) to human vascular endothelial and smooth muscle cells. Blood 1997; 89(12): 4290–4298.
21. Philips ML, Lembertas AV, Schumaker VN et al. Physical properties of recombinant apo(a) and its association with LDL to form an Lp(a)-like complex. Biochemistry 1993; 32(14): 3722–3728.
22. Yano Y, Shimokawa K, Okada Y et al. Immunolocalizacion of lp(a) in wounded tissues. J Histochem Cytochem 1997; 45(4): 559–568. doi: 10.1177/ 002215549704500408.
23. Baggio G, Donazzan S, Monti D et al. Lipoprotein(a) and lipoprotein profile in healthy centenarians: a reappraisal of vascular risk factor. FASEB J
1998; 12(6): 433–437. doi: 10.1096/ fasebj.12.6.433.
24. Marcovina SM, Albers JJ, Wijsman E et al. Differences in Lp(a) concentrations and apo(a) polymorphysm between black a white Americans. J Lipid Res 1996; 37(12): 2569–2585.
25. Kojima S, Harpel PC, Rifkin DB et al. Lipoprotein (a) inhibits the generation of trasforming growth factor beta: an endogenous inhibitor of smooth muscle cell migration. J Cell Biol 1991; 113(6): 1439–1445. doi: 10.1083/ jcb.113.6.1439.
26. Heberland ME, Fless GM, Scanu AM et al. Malondialdehyde modification of lipoprotein(a) produces avid uptake by human monocytemacrophages. J Biol Chem 1992; 267(6): 4143–4151.
27. Nachman RL. Thrombosis and atherogenesis: molecular connections. Blood 1992; 79(8): 1897–1906.
28. Hajjar KA, Nachman RL. The role of lipoprotein(a) in atherogenesis and thrombosis. Annu Rev Med 1996; 47: 423–442. doi: 10.1146/ annurev.med.47.1.423.
29. Cooke JP. The pathophysiology of periferal arterial disease: rational targets for drug intervention. Vasc Med 1997; 2: 227–230.
30. Seman LJ, DeLuca C, Jenner JL et al. Lipoprotein(a)-cholestrol and coronary heart disease in the Framingham Heart Study. Clin Chem 1999; 45(7): 1039–1046.
31. Milionis HJ, Winder AF, Mikhailidis DP et al. Lipoprotein(a) and stroke. J Clin Pathol 2000; 53(7): 487–496. doi: 10.1136/ jcp.53.7.487.
32. Miner SE, Hegele RA, Sparkes J et al. Homocystein, lipoproteine(a) levels and restenosis after PTCA: a prospective study. Am Heart J 2000; 140:
272–278.
33. von Depka M, Nowak-Göttl U, Eisert R et al. Increased lipoprotein (a) levels as an indipendent risk factor for venous tromboembolism. Blood 2000; 96(10): 3364–3368.
34. Angelin B. Therapy for lowering lipoprotein (a) levels. Curr Opin Lipidol 1997; 8(6): 337–341.
35. Seed M, O’Connor B, Perombelon N et al. The effect of nicotin acid and acipimox on lipoprotein (a) concentration and turnover. Atherosclerosis 1993; 101(1): 61–68.
36. Ramharack R, Spahi MA, Hicks GW et al. Gemfibrozil significantly lowers cynomolgus monkey plasma lipoprotein (a)-protein and liver apolipoprotein (a) mRNA levels. J Lipid Res 1995; 36(6): 1294–1304.
37. Galetta F, Sampietro T, Basta G et al. Effects of simvastatin on blood levels of lipoprotein (a). Minerva Med 1995; 86(7–8): 299–303.
38. Bambauer R, Schiel R, Keller HE et al. Low-density lipoprotein apheresis in the treatment of two patients with coronary heart disease and extremely elevated lipoprotein (a) levels.. Artif Organs 1996; 20(4): 340–343.
39. Henriksson P, Angelin B, Berglund L. Hormonal regulatulation of serum Lp (a) levels. Opposite effects after estrogen treatment and orchidectomy in males with prostatic carcinoma. J Clin Invest 1992; 89(4): 1166–1171. doi: 10.1172/ JCI115699.
40. Navarese EP, Kolodziejczak M, Schulze V et al. Effects of proprotein convertase subtilisin/ kexin type 9 antibodies in adults with hypercholesterolemia: a systematic review and meta-analysis. Ann Intern Med 2015; 163(1): 40–51. doi: 10.7326/ M14-2957.
41. Gaudet D, Watts GF, Robinson JG et al. Effect of alirocumab on lipoprotein(a) over ≥1.5 years (from the Phase 3 ODYSSEY Program). Am J Cardiol 2017; 119(1): 40–46. doi: 10.1016/ j.amjcard.2016.09.010.
42. O'Donoghue ML, Fazio S, Giugliano RP et al. Lipoprotein(a), PCSK9 inhibition, and cardiovascular risk. Circulation 2019; 139(12): 1483–1492. doi: 10.1161/ CIRCULATIONAHA.118.037184.
43. Viney NJ, van Capelleveen JC, Geary RS et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 2016; 388(10057): 2239–2253. doi: 10.1016/ S0140-6736(16)31009-1.
44. Catapano AL, Graham I, De Backer G et al. 2016 ESC/ EAS Guidelines for the management of dyslipidaemias. Eur Heart J 2016; 37(39): 2999–3058. doi: 10.1093/ eurheartj/ ehw272.
Štítky
Dětská kardiologie Interní lékařství Kardiochirurgie KardiologieČlánek vyšel v časopise
Kardiologická revue – Interní medicína

2019 Číslo 2
Nejčtenější v tomto čísle
- Betablokátory u kardiovaskulárních onemocnění – pro a proti
- Update role Lp (a) při určení kardiovaskulárního rizika a možnosti jeho ovlivnění
- Minimum o mechanických podporách srdca – klasifikácia, indikácie, princípy, klinické skúsenosti
- Novinky o familiární hypercholesterolemii pro kardiology